How Much Can I Inject? Part II: Injecting in Solvents Other than Mobile Phase
LCGC North America
How large an injection can you make if the injection solvent is not matched to the mobile phase?
How large an injection can you make if the injection solvent is not matched to the mobile phase?
In last month's "LC Troubleshooting" discussion (1), we looked at the effect of the injection volume on the increase in peak width or reduction in resolution when the sample was injected in the mobile phase for a liquid chromatography (LC) separation. We saw that the allowed injection volume was related to the volume of the peak generated by the column in the absence of injection-related effects. Columns that generated smaller peak volumes required smaller injection volumes or the peaks began to deteriorate. Smaller peak volumes result from any combination of a reduction of column length, diameter, or packing particle diameter. Thus, the move from traditional 150 mm × 4.6 mm, 5-μm particle diameter (dp) columns to 100 mm × 2.1 mm, 3-μm dp columns to 50 mm × 2.1 mm, 1.7-μm dp columns resulted in smaller and smaller peak volumes as well as smaller recommended injection volumes. We distilled all this information into an easy-to-remember rule of thumb, the 15% Rule, which reminds us that as long as we keep mobile phase as the injection solvent and inject no more than 15% of the volume of the first peak of interest, we should observe no more than a 1% increase in peak width or a 1% decrease in resolution. Larger injection volumes than recommended by the 15% Rule will result in greater increases in peak width and subsequent decreases in resolution. Examples were presented for all these cases.
This month, we look at what happens when we deviate from the above recommendations. Specifically, what happens if we use an injection solvent that is stronger or weaker than the mobile phase? After all, many times the sample comes to us in something other than the mobile phase, and we'd rather not add sample preparation steps to transfer the sample to a new solvent. Or sometimes the sample is too dilute to see an adequate signal if we stick to the 15% Rule — is there a way to increase the injection volume while mitigating unwanted peak broadening? All the examples so far have been for isocratic separation — how does it work for gradients?
The Effect of Retention
Last month (1), we looked at the influence on allowable injection volume for a variety of column configurations and injection volumes for a peak with a retention factor (k) of 1. This is a logical approach, because usually the first peak in the separation is the one most influenced by the injection. This month, I'm going to restrict the discussion to a single column: 100 mm × 4.6 mm, packed with 3-μm dp particles and operated at 1 mL/min. This column will have a dead volume, Vm, of approximately 1 mL, which generates a dead time, t0, of 1 min at a flow rate of 1 mL/min. For a real sample with a routine method, we can expect a column plate number, N, of approximately 10,000 plates under these conditions. The examples under these conditions are easily transposed to the other columns discussed last month with similar conclusions related to the change in column size.


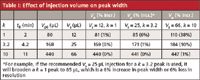
Table I: Effect of injection volume on peak width
First, let's look at the allowed injection volume for a peak of a given retention. In Table I, I've listed in the left-hand column k values of 1, 3.2, and 10 for three peaks and next to them their corresponding retention times under the conditions listed above. Our column will generate peak widths that increase with retention times, because


where tR is the retention time and w is the peak width at baseline between tangents drawn to the sides of the peak. For the present discussion, we're interested in the injection volume of the sample, Vs, so we need to convert equation 1 into volume units by changing retention time into retention volume, (VR = tR × F, where F is the flow rate), and peak width from time to volume (= w × F). We'll start with the theoretical peak volume, Vp0, in the absence of any injection or other extra-column peak broadening. Because N should be constant for all peaks in an isocratic separation, as tR or VR is increased, w or Vp0 must increase to keep equation 1 balanced. The peak volume is listed in the third column of Table I.
I won't repeat all the equations from last month's discussion (1), but the calculation of the observed peak volume is useful (this was equation 4 in the earlier discussion):

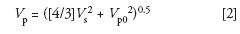
where Vp is the observed, broadened peak.
Last month we learned, as summarized in the 15% Rule, that we could inject up to 15% of the volume of a peak without broadening it more than 1%. Remember that any increase in peak width is directly related to a corresponding loss in resolution between two adjacent peaks, so this injection volume, Vs, would cause no more than a 1% loss in resolution. I have shown the allowed injection volumes using the 15% Rule for each peak in the fourth column of Table I. The right-hand portion of Table I shows the effect of using these injection volumes. Let's look first only at the k = 1 and k = 10 peaks. For the k = 1 peak, if I inject the recommended Vs = 12 μL, the resulting observed peak width, Vp, is 81 μL, which corresponds to a loss of 1% in resolution over the original Vp0 = 80 μL peak, as shown by the data in the fifth column of Table I on the row corresponding to k = 1. (As usual, I've rounded or truncated numbers in the tables for display purposes, so if you repeat these calculations, your results will vary slightly.) In the same way, the k = 10 peak has a maximum recommended injection of Vs = 66 μL, which generates a final peak volume, Vp = 447 μL, also an increase of 1% (last column and row of Table I).
It should be obvious that longer-retained peaks are allowed larger injection volumes because of their larger inherent peak widths, as illustrated above for Vs of 12 or 66 μL for the peaks with k values of 1 and 10, respectively. This would be a simple application if the two peaks were in separate chromatograms, but what happens if the two peaks are in the same sample? If we choose the injection volume for the k = 1 peak, because it is fivefold smaller than the k = 10 recommendation, we would expect there to be little or no broadening of the k = 10 peak. This is observed for data in the last row (k = 10) and fifth column (Vs = 12 μL) of Table I. You can see that the final peak volume Vp = 440 μL is the same as the unbroadened peak, Vp0 for k = 10. On the other hand, if we choose the injection volume recommended for the larger-volume peak, the results can be disastrous. You can see what will happen if the Vs = 66 μL is applied to the k = 1 peak (upper right data point in Table I) — the peak is broadened by 38%. This is why, when we apply the 15% Rule, we only consider the peak volume of the first peak of interest — it is the most sensitive to injection volume.
The Effect of a Stronger Injection Solvent
Another way of looking at the data in Table I will help us understand the influence of the injection solvent strength on the chromatogram. Let's consider a case where the injection solvent is stronger than the mobile phase. By stronger, I mean that the percent organic solvent (%B) is higher in the injection solvent than the mobile phase. We can estimate the change in retention, Δk, for a change in %B (Φ = 0.01 × %B, so Φ = 0.1 ≡ 10% B) with

where S is a characteristic of each analyte, estimated as S ≈ 0.25 MW0.5, where MW is the molecular weight. If we choose a typical 400 Da small molecule, S ≈ 5 and Δk ≈ 3.2 for a 10% change in %B. This means that a 10% increase or decrease in the mobile phase %B will result in a corresponding threefold reduction or increase, respectively, in the k values. The effect on retention time is approximately the same, because k = (tR – t0)/t0. This result is the source of the Rule of Three often mentioned in "LC Troubleshooting." A change in %B of 20% will result in approximately a 10-fold change in k values; making a generic estimate of the change in retention for even larger changes in %B will likely be less useful because of the assumptions inherent in equation 3.
If sample is injected in a solvent that is stronger than the mobile phase, for the time that the analyte molecules are in that injection solvent before it gets diluted by mobile phase, the injection solvent effectively is the mobile phase. As the plug of injection solvent moves through the column, it will move at the same flow rate as the mobile phase, but the sample molecules in it will move more quickly than they would in mobile phase because the "mobile phase" they see is the injection solvent. The injection plug, of course, will get diluted with mobile phase until it is no longer distinguishable from the mobile phase. The mobile phase following the injection plug will dilute it, so the molecules at the tailing edge of the injection plug will slow and travel at the normal elution rate before those on the front edge. Although all this happens quite quickly, there can be significant band broadening in the process if the solvent is strong enough and the volume is large enough.
I've shown the process conceptually in Figure 1. Note that this figure is oversimplified, but it should serve to illustrate what happens. In each case, I've shown a portion of the column as a horizontal rectangle filled with mobile phase (dot fill). In the top three examples, sample injected in mobile phase is shown as the checkerboard pattern. As time passes from A to B to C, the sample moves through the column in the normal manner, with some band broadening (not shown) taking place. In the bottom three examples, the sample is dissolved in a stronger solvent (diagonal fill). In A', right after injection, the sample takes up the same amount of space at the top of the column because it is the same injection volume as in A. However, in B', because the injection plug is a stronger solvent than the mobile phase, the molecules in the plug travel more quickly through the column than normal. At the same time, the back edge gets diluted with mobile phase, so those analyte molecules slow down to the normal migration rate (checkerboard pattern). Even though the injection plug is narrower now, because it is becoming diluted at the front and back edges, the sample is spread over a larger portion of the column. In C', the injection plug is mostly diluted, but the spreading continues to take place until all the sample is in mobile phase (not shown), at which point the now broadened band travels in the normal manner. You can get a visual concept of why the injection band gets broadened when a stronger solvent is used: The front edge of the peak gets pushed forward relative to its expected behavior in mobile phase, whereas the back edge of the peak travels at the same speed as if it were injected in mobile phase.

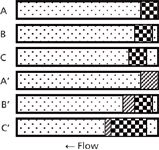
Figure 1: A, B, and C illustrate the concept of a sample plug injected in mobile phase as the injection solvent as it moves through the column (right to left). A’, B’, and C’ illustrate the same process with the use of the same volume of a stronger injection solvent. Mobile phase, dot pattern; sample in mobile phase injection solvent, checkerboard; sample in stronger injection solvent, diagonal pattern. See text for details.
One solution to this excessive broadening during injection would be to use a less-strong injection solvent. The more different the injection solvent is from the mobile phase, the more severe the problem will be. As we saw above for normal band migration, a 10% increase in %B will cause sample to move about three times faster than normal and a 20% change will increase that rate to approximately 10-fold. Another solution would be to inject less sample. The smaller the injection plug volume, the faster it will get diluted into the mobile phase. I remember one case in our laboratory where we were able to inject a 1-μL volume of sample dissolved in toluene into a reversed-phase mobile phase and get satisfactory results. Although the toluene was nominally immiscible, the small volume allowed it to dissolve or disperse in the mobile phase instantly before any noticeable band migration had occurred.
What About a Weaker Injection Solvent?
When the injection solvent is weaker than the mobile phase, the sample molecules initially slow down relative to their normal mobile-phase migration rates. This process often is referred to as "band compression," or "on-column concentration." Using the above examples of a 10% and 20% change in %B, we would expect the sample to slow its migration to approximately 1/3 and 1/10 of the normal rates, respectively. In the extreme, such as injecting a 100% aqueous sample into a 50% or more organic solvent mobile phase, the sample effectively stops at the top of the column until a strong enough solvent (the mobile phase) comes along to cause it to migrate normally. For example, if you were trying to analyze for trace contaminants in river water, you might be able to inject 10 mL or more of sample without adverse effects, increasing the injected sample mass without having to go through preconcentration steps during sample preparation.
And Gradients?
The same processes that we've looked at above for isocratic separations take place with gradients. The main difference is that usually the initial gradient conditions consist of a very weak mobile phase, often 5–10% B. This can severely restrict the allowable organic concentration of the mobile phase. On the other hand, because the mobile phase is so weak, it will be much more effective at diluting a stronger injection solvent than an isocratic mobile phase of 50% or 60% B, so the use of a stronger injection solvent may be mitigated somewhat.
So, How Do I Tell?
You'll notice that I've done some hand-waving while trying to explain what happens when the sample is injected in a solvent other than the mobile phase. This is because there are several processes going on simultaneously that are not as easy to model as the case of mobile phase as the injection solvent. This situation is complicated by how much band broadening or loss of resolution you can tolerate for a given application. Further confusing things is the injection process itself — band broadening can vary with different autosampler designs. In any event, I suggest using an empirical approach to address the selection of any injection volume, even in mobile phase. This approach works both for isocratic and gradient methods.
First, use the 15% Rule or one of the tables in last month's discussion (1) to get an idea of how much you can inject. Round the suggested volume to something convenient. For example, rather than the 12 μL recommendation of Table I, select 10 μL or 15 μL. Second, inject this volume, then inject half the volume and twice the volume. Finally, compare the chromatograms and see what is acceptable. If the half-size injection gives much narrower peaks with better resolution, perhaps you should reduce the injection volume and test it with the ±2× volume test. If the larger injection still looks good, maybe you can inject an even larger volume. Because you want robust conditions that won't cause problems when some unexpected change happens, it is a good idea to leave a safety margin in the chosen injection volume. For example, if 20 μL looks good, but 40 μL shows problems, check 30 μL. If this still looks acceptable, you'll probably be safe with 20 μL, but if 25 μL shows problems, you probably are working too close to the edge of reliability.
In summary, I can think of no better advice than that of I.M. Kolthoff, often referred to as the father of analytical chemistry, who said, "Theory guides, experiment decides." We can spend lots of time with equations, and they are helpful at getting a good idea of what should happen, but there is nothing like a well-designed experiment to see if the results are acceptable.
References
(1) J.W. Dolan, LCGC North Am. 32(10), 780–785 (2014).
John W. Dolan "LC Troubleshooting" Editor John Dolan has been writing "LC Troubleshooting" for LCGC for more than 30 years. One of the industry's most respected professionals, John is currently the Vice President of and a principal instructor for LC Resources in Walnut Creek, California. He is also a member of LCGC's editorial advisory board. Direct correspondence about this column via e-mail to John.Dolan@LCResources.com


John W. Dolan











Investigating the Protective Effects of Frankincense Oil on Wound Healing with GC–MS
April 2nd 2025Frankincense essential oil is known for its anti-inflammatory, antioxidant, and therapeutic properties. A recent study investigated the protective effects of the oil in an excision wound model in rats, focusing on oxidative stress reduction, inflammatory cytokine modulation, and caspase-3 regulation; chemical composition of the oil was analyzed using gas chromatography–mass spectrometry (GC–MS).


Evaluating Natural Preservatives for Meat Products with Gas and Liquid Chromatography
April 1st 2025A study in Food Science & Nutrition evaluated the antioxidant and preservative effects of Epilobium angustifolium extract on beef burgers, finding that the extract influenced physicochemical properties, color stability, and lipid oxidation, with higher concentrations showing a prooxidant effect.

Rethinking Chromatography Workflows with AI and Machine Learning
April 1st 2025Interest in applying artificial intelligence (AI) and machine learning (ML) to chromatography is greater than ever. In this article, we discuss data-related barriers to accomplishing this goal and how rethinking chromatography data systems can overcome them.


