How Do You Write User Requirements for Chromatographs and Chromatography Data Systems?


One of the biggest failures with purchasing chromatograph systems and chromatography data system (CDS) software is either the total lack of or poorly written user requirements. So, how can you write acceptable requirements? Is specifying a chromatograph the same as software?
One of the biggest failures with purchasing chromatograph systems and chromatography data system (CDS) software is either the total lack of or poorly written user requirements. So, how can you write acceptable requirements? Is specifying a chromatograph the same as software?
When it comes to the purchase of chromatographs or chromatography data system (CDS) software, the worst possible task for a user is to specify what they want it to do. Users either “can’t be bothered” or “know what they want”. With chromatographers like this, the world will always need consultants, if not to help them do the job properly in the first place then to dig them out of the hole that they dug themselves. In this instalment of “Questions of Quality” the writing of a user requirements specification (URS) for both a liquid chromatograph system and CDS software is discussed.
Why Did You Buy This Rubbish?
Be honest, have you ever bought a chromatograph system that was an absolute lemon or CDS that failed to meet your expectations? I have. This column is written for
all those readers who lied when answering the question in the first sentence. Let’s look at some of the miserable excuses for this sorry state of affairs:
- It’s a chromatograph-it works
- I was too busy
- I was too busy so I went with my friend’s suggestion
- I was never given time by my boss
- I believed the supplier’s literature
- The salesman bought me a nice meal and it would be impolite not to order
- My boss said we must spend the money before the end of the month or lose it.
The root cause of this is the abject failure to plan and make the time available to specify your requirements adequately for instruments and software. With an adequate URS you can evaluate the software or chromatograph objectively. You only have one chance to get a purchase right, otherwise you’ll have to live with your lemon for several years. Amazon returns are not available for chromatograph systems or CDS software.
The way out of this quagmire is to write meaningful user specifications that will enable you and your laboratory to spend money wisely and get the right instrument and CDS for the job. There is a caveat: buying only on price can be a false economy in the long run.
Why Do We Need Specifications?
There are two main reasons for writing user specifications.
1. Investment protection: You want the right tool for the right job. Buying the wrong item will give you more problems over the lifetime of the instrument than spending the time to write down what you want in the first place. Buying the wrong item wastes scarce resources and makes you look an idiot with management.
2. Compliance with regulations or quality standards: The laboratory or organisation is required to do this to meet their legal requirements or quality commitments.
You may think that these are two entirely different areas but you are wrong. If you approach the writing of user requirements with a business-driven attitude but with a compliance or quality wrapper, you can kill the two proverbial birds with one stone. If you write down your requirements with adequate document controls and approve them, then this meets both reasons for writing specifications. Note, I mentioned the business rationale for writing requirements first as this must be the main driver for writing a URS.
Further Business Rationale for Writing a URS
Don’t forget the real reasons for writing a URS, especially for CDS software, are the following (1):
- It serves as a reference against which commercial products are selected, evaluated in detail, and any enhancements are defined. You avoid being seduced by technology or buying a poor system using this approach.
- It identifies gaps between your requirements and the CDS applications offered by suppliers. This allows you to seek enhancement of the selected system or to review and possibly adjust your requirements to match software on the market.
- It reduces the total system effort and costs, since careful review of the document should reveal omissions, misunderstandings, or inconsistencies in your specification and this means that they can be corrected easily before you purchase an instrument or application.
- It provides the input to user acceptance test specifications and qualification of the system.
- It defines what procedures need to be written, modified, or archived for both the users and administrators of the system.
You will have noticed that I have not mentioned any regulations or quality guidelines, merely described what has happened in many laboratories when chromatograph systems and software are purchased. However, I don’t wish to disappoint you, so here are the quality standard requirements and pharmaceutical regulations you may need to consider.
ISO 17025 Quality Standard
ISO 17025: 2005, a quality standard for testing and calibration laboratories, presents in Section 5.5 the following requirements for analytical instruments:
5.5.2 Equipment and its software used for testing, calibration, and sampling shall be capable of achieving the accuracy required and shall comply with specifications relevant to the tests and/or calibrations concerned.…
Before being placed into service, equipment (including that used for sampling) shall be calibrated or checked to establish that it meets the laboratory’s specification requirements and complies with the relevant standard specifications (2).
Note the highlighted text “laboratory’s specification requirements”. Not the supplier’s but the laboratory’s specification. This implies that there can be a difference between the supplier’s specification and that required by the laboratory. The important point is that a laboratory does not have to follow the supplier’s specification to the letter; the key point is what does the laboratory want an instrument to do?
The 2017 update of ISO 17025 (3) has omitted this requirement and simply asks in section 6.4.1 for access to equipment ... that is required for the correct performance of laboratory activities ... It is a pity that guidance for laboratory specifications has been lost from this standard.
Good Laboratory and Manufacturing Practice Regulations
Let us move onto US Good Laboratory Practice (GLP) and Good Manufacturing Practice (GMP) Regulations for Equipment and see what they say about specifications. The US GMP Regulations for Equipment are found in section 21 CFR 211.63 (4) and state:
Equipment used … shall be of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and for its cleaning and maintenance.
The corresponding regulations for GLP are in section 21 CFR 58.61 (5) and are as follows:
Equipment used … shall be of appropriate design and adequate capacity to function according to the protocol and shall be suitability located for operation, inspection, cleaning and maintenance.
Both US GMP and GLP require appropriate design suitable for intended use or function for the protocol, respectively. Intended use has been interpreted as documenting requirements, otherwise how can you determine what the use will be and verify that it works? However, both of these regulations were originally drafted in the 1970s and are relatively vague-is there anything more up-to-date and informative?
General Principles of Software Validation
If you think that all this writing requirements is rubbish and a waste of time, Section 5.2.2 of this FDA Guidance states simply (6):
It is not possible to validate software without predetermined and documented software requirements.
Now that I have your undivided attention, please read on.
USP <1058> Analytical Instrument Qualification (AIQ)
The latest version of USP <1058> on Analytical Instrument Qualification has the following statements (7):
The first activity is the generation of a user requirements specification (URS), which defines the laboratory’s particular needs and technical and operational requirements that are to be met.
The subsequent qualification activities necessary to establish fitness for purpose may be grouped into four phases: design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ).
Therefore, writing the URS for an analytical instrument is a totally separate activity than the design qualification (DQ) phase or selecting the instrument and supplier.
DQ is an activity that confirms that the instrument(s) you propose purchasing meet the requirements in your URS. This is consistent with requirements 3.2 and 3.3 in EU GMP Annex 15 on Qualification and Validation (8). You test the user requirements in the operational qualification (OQ) phase of work. This was discussed in past instalments of “Questions of Quality” focused on the new version of USP <1058> (9) and in the three papers co-authored with Paul Smith (10–12).
The problem now comes with a further statement in the new USP <1058>:
It is expected that DQ requirements will be minimal for commercial, off-the-shelf instruments.
Note these words well: requirements will be minimal. Not zero, not virtual, not imaginary, not verbal, but minimal. Minimal means written down, documented, recorded, and formal (approved).
Minimal Instrument Requirements But What About Software?
While instrument specifications may be minimal, this is certainly not the case for software. Here the functions need to be specified for instrument control, data acquisition, processing, calculation of results, and reporting. CDS applications are configurable and the configuration needs to be documented as either part of the overall specification, for example, system policies, user roles, use of electronic signatures. Alternatively, more dynamic aspects of the system, such as specifying, building, testing, and maintenance of custom calculations and reports, should be controlled by procedure. This is not an exercise in minimalism.
Different Approaches for Chromatograph and CDS Specifications?
From the discussion above, we appear to have a dichotomy with our URS documents. On the one hand the chromatograph specification is expected to be minimal, but should be much more detailed for the CDS application software. To help you with this crucial task we’ll have a look at practical approaches to specifying both components. We’ll start with our exercise in minimal high performance liquid chromatography (HPLC) user requirements. For many, the first response is to quote the supplier’s specification verbatim.
Why Can’t I Use a Supplier’s Specification?
Yes, I know you are lazy and have analyses to perform, but this is not the way to write your specification. There are several reasons for this:
A supplier’s specification covers the entire operating range of the instrument. Do you really want an HPLC pump specification to cover the range of 0.05–10.0 mL/min, when you only use 1.0–2.0 mL/min? If you specify the whole range, you will have to test it to show it complies. Similarly, a UV detector may have an operating range from 190–800 nm. What do you use normally? Perhaps 210–280 nm, for example? Focus on what you actually need now rather than what you could use. I know there are those that suggest a wider operating range-just in case. However, resist this suggestion and use change control once the instrument is operational to adjust your approach. You know it makes sense.
A supplier’s specification will have operating parameters measured under highly-controlled environmental conditions that your laboratory cannot hope to match. Therefore USP <1058> wants suppliers to generate meaningful specifications (7) so that they can be reproduced in customers’ laboratories.
What Could a Minimal HPLC Specification Look Like?-Isocratic HPLC
An example of a simplified and minimal specification for an isocratic HPLC is shown in Table 1. It details a supplier’s operating range for each component in the middle column and then in the right-hand column are the laboratory’s requirements, which are selected from the supplier’s operating range. It took me about five minutes to write this outline specification. It’s not that hard to write a specification, is it?


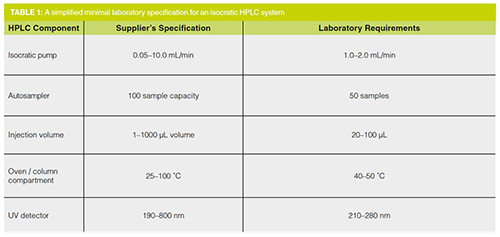
Hold on, is there something missing from this specification? Of course, the acceptance criteria for each parameter are missing and these are an integral part of any laboratory instrument specification. Otherwise, how can you test or qualify a component to demonstrate that it is fit for intended use?
How should these acceptance criteria be derived?
1. The overriding requirement is that any acceptance criterion must be scientifically sound as required by 21 CFR 211.160(b) (4) and ICH Q7(R1) clause 11.1 (13).
2. There are acceptance criteria for many analytical instruments in the general chapters of the pharmacopoeias.
One piece of advice I would offer is use the pharmacopoeial acceptance criteria as written and not to make them tighter. They have been specified for a reason following discussion and debate across industry.
What Could a Minimal HPLC Specification Look Like?-Gradient HPLC
Table 1 shows the simplified specification for an isocratic HPLC. What would happen if you wanted a gradient chromatograph? How would you specify this? For example, you could have a simple binary system or would you want a quaternary gradient system? Let’s assume the Gods of Finance have been kind and bestowed upon you the cash to splash on a quaternary system. How do you envision using the system? I appreciate the hotshots in R&D are itching to develop a quaternary gradient separation to show off their superior chromatography skills to the mere mortals in the quality control department, however, let’s get real. To have a robust method remember the KISS principle: keep it simple, stupid.
When developing a method the principle should always be isocratic separation first, gradient separation second. If a gradient separation is required, we should use a binary system and not a tertiary or a quaternary system. How do we normally use a quaternary HPLC pump? Typically, A and B will be the solvents for a binary gradient, C will be an aqueous wash, and D will be an organic wash such as methanol or acetonitrile. In our minimal specification we need to state this. Consider what acceptance criteria would you want. Obviously, you’ll need to look at the accuracy of mixing A and B solvents along with the overall performance of the mixed mobile phase flow rate accuracy. However, do you need to specify any acceptance criteria for solvents C and D? If you take a risk-based approach, probably not. All done?
Not quite, how would you mix the gradient? Low or high pressure mixing? Does it really matter? Yes, it does, especially if you are transferring a method from one laboratory to another because how the gradient is mixed could potentially impact a separation. If one laboratory has low pressure mixing and the other high, there could be problems reproducing the original gradient.
The outline specification shown in Table 1 is the start of the specification journey, but you can see that it is not a difficult task to develop a meaningful but minimal specification for a chromatograph system with acceptance criteria. Each parameter can be tested objectively for each module if required, but don’t forget that a holistic test to demonstrate that the whole chromatograph system works is also required (14).
What About Software Specifications for a CDS?
We have looked at how specifications for commercial instruments are expected to be minimal for a liquid chromatograph system. Now we need to ask the same question for software. The answer for any chromatography data system is, quite simply, no. The reason is the difference between a software application vs. an instrument. An instrument, in our case a chromatograph, is relatively simple, with some of the operating parameters outlined in Table 1.
CDS application software is much more complex and its impact is far greater: it can control a single chromatograph system in a single laboratory or multiple systems in multiple sites globally. The URS scope applies for a standalone system as well as a global one. Rather than have a small set of operating parameters, a CDS application has a wide range of functions such as:
- Instrument control
- Sequence file to identify the injections to be made and input of factors such as dilutions, weights, purities, and water content of standards
- Data acquisition
- Automatic and manual integration
- Calculations such as system suitability parameters and the reportable result.
In addition, a CDS also needs to be configured and this must be documented:
- Log on security, for example, password length and complexity
- User roles and access privileges that avoid conflicts of interest
- Unique user identities
- Protection of electronic records, for example, restriction of deletion privileges, audit trail turned on
- Implementation of electronic signatures
- Defining data storage locations
- Custom calculations
- Custom reports.
And there’s more requirements:
- Time synchronisation
- Cyber security
- System patching, for example, security patches
- Change control and configuration management
- Backup and recovery
- Archive and restore
If the system is supported by an IT group there also needs to be an agreement of the services provided, and the roles and responsibilities of the laboratory and IT provider as required by EU GMP Chapter 7 and Annex 11 (15,16).
Scope of a URS for a CDS
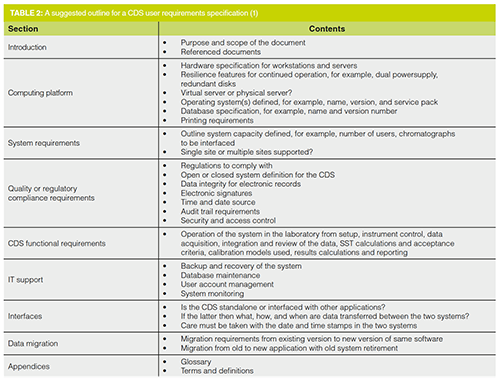
The areas listed above need to be arranged into groups of similar requirements. One such way of doing this is presented in Table 2. You can immediately contract this with the minimal requirements for the chromatograph shown in Table 1, the difference is simply the wider scope and complexity needed to adequately define the requirements for a CDS.

Guidance for Writing User Requirements
The following general guidelines should be followed during the writing of the CDS URS:
- Each requirement statement must be uniquely referenced.
- Do not use the word processor auto-numbering function for requirement numbering. If a new requirement is added all subsequent ones are incremented and traceability will be lost. You have been warned.
- Each requirement should be short (typically no longer than 25 words).
- Each requirement should only contain one requirement otherwise it is difficult to trace.
- The URS should be consistent and requirements should not be duplicated or contradicted.
- Specify requirements and not design solutions. The focus should be on what is required, not how it is to be achieved.
The exception to the point above is where corporate IT standards become a constraint on the system, for example, when a specific database or operating system must be used and no others are allowed
Each requirement should be testable or verifiable. Testable is defined as test cases can be derived from the requirement as written. This allows the tests to be designed as soon as the URS is finalised. A requirement can be verified through an activity (for example, IQ of a component or writing of a standard operating procedure [SOP] verifies that the requirement has been met).
Both the laboratory and the supplier must understand the document. Jargon should be avoided wherever possible and key words are defined in a specific section in the document.
Requirements should be prioritised. There are various schemes that could be used but I prefer simplicity and typically use mandatory (essential to meet business or regulatory requirements) or desirable (nice to have).
The URS should be modifiable, but changes should be under a formal control procedure. The easiest is by up-versioning and authorising the new version then archiving the old document.
A URS is correct if every stated requirement has only one interpretation and this is met by the system. Unfortunately, this is very rare.
Who Should be Involved in Writing a URS?
Writing a user requirements specification for a CDS is not difficult, but the process is not a trivial exercise. It requires the involvement of a multidisciplinary team to write a URS consisting of chromatographers, quality, and, if the system is networked, IT. You will notice that there is no role for a supplier. That is because you have not selected the CDS yet and you are writing a generic specification.
After selection you will need to update the document to make it specific for the chosen application (name and version number) and here the supplier can help with training key users and a review of the updated document.
How to Write Testable or Verifiable User Requirements
This is the heart of a good or bad URS. If you can’t test or verify a requirement, it is of zero value. Meaningless requirements may impress management but they don’t define the intended use of the instrument or software. To illustrate some of the problems of writing testable user requirements, here are two examples of how not to write requirements for a CDS. Note that both requirements are uniquely numbered, which is good, but these are real examples, which is not.
Performance requirement: 6.1.8: Operating at normal PC response times with no undue delay in response at low computer utilisation.
In requirement 6.1.8 there is wording that makes the requirement untestable. These are typically called weasel words, which are normal, undue, and low. These render the requirement useless and incapable of being tested. For example, what is a normal PC response time and what is undue delay? These are meaningless and untestable words.
A further example is shown below.
Reporting 6.2.4: Report production at a rate of at least a page every 10 seconds at modest network and server utilisation.
Requirement 6.2.4 is marginally better as “report production at a rate of at least one page every 10 seconds” is testable and specific. However, the requirement then snatches defeat from the jaws of victory with the phrase “at modest network speed”, rendering it untestable as “modest” cannot be defined.
For those readers that love simplicity when writing requirements there is the ever-popular:
4.2.1: The system must be 21 CFR 11 compliant.
When I read such a requirement I do not know if it has been written by a stupid or a lazy person, or both. The writer does not understand that the 21 CFR 11 regulation is divided into technical, procedural, and administrative requirements.
A URS is a Living Document
It is vital to understand that the contents in a URS are not static. As your chromatographic needs change so too may your CDS and chromatograph requirements. As a simple example, if your UV detector is qualified between 210 nm and 280 nm and a new analyte method has detection at 310 nm, then you need to update the instrument specification and requalify the detector. Similarly, if you change your working practice and implement electronic signatures, then the URS, configuration settings, and testing documents all need to be updated. In regulated laboratories there must be change control that examines the impact of a change on instruments, CDS software, and documentation including specifications and procedures.
Summary
We have considered what appears to be one of the most difficult tasks in the laboratory: writing effective user requirements for chromatograph systems and chromatography data system software. It is not an arduous task but requires time that management must realise and allow for. One example I saw in an audit consisted of six requirements and 13 words that were only written to keep quality assurance (QA) happy. It may keep QA quiet but it will not impress auditors and inspectors. Improvement of user requirements specifications is a key component of continual improvement in any quality system.
References
- R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements, Second Edition (Royal Society of Chemistry, Cambridge, UK, 2017).
- ISO 17025, General requirements for the competence of testing and calibration laboratories (International Standards Organization, Geneva, Switzerland, 2005).
- ISO 17025, General requirements for the competence of testing and calibration laboratories (International Standards Organization, Geneva, Switzerland, 2017).
- 21 CFR 211, Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration: Silver Spring, Maryland, USA, 2008).
- 21 CFR 58, Good Laboratory Practice for Non-Clinical Laboratory Studies (Food and Drug Administration, Washington, D.C., USA, 1978).
- Food and Drug Administration, FDA Guidance for Industry General Principles of Software Validation (Food and Drug Administration, Rockville, Maryland, USA, 2002).
- General Chapter <1058> “Analytical Instrument Qualification”, in United States Pharmacopeia 41 (United States Pharmacopeial Convention, Rockville, Maryland, USA, 2018).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15 Qualification and Validation (European Commission, Brussels, Belgium, 2015).
- R.D. McDowall, LCGC Europe31(1), 36–41 (2018).
- P. Smith and R.D. McDowall, LCGC Europe31(7), 385–389 (2018).
- P. Smith and R.D. McDowall, LCGC Europe31(9), 504–511 (2018).
- P. Smith and R.D. McDowall, LCGC Europe32(1), 28–32 (2019).
- International Conference on Harmonization, ICH Q7(R1) – Good Manufacturing Practice for Active Pharmaceutical Ingredients (ICH, Geneva, Switzerland, 2016).
- W.B. Furman, T.P. Layloff, and R. Tetzlaff, JOAC International77, 1314–1317 (1994).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 11 Computerised Systems (European Commission, Brussels, Belgium, 2011).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 7 Outsourced Activities (European Commission, Brussels, Belgium, 2013).
“Questions of Quality” editor Bob McDowall is Director of R.D. McDowall Limited, Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column to the editorâinâchief, Alasdair Matheson, amatheson@mjhlifesciences.com







What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






