Highlights of HPLC 2008: Part II
LCGC North America


Ron Majors continues his summary of the technical highlights of HPLC 2008, discussing more hot topics from the show.
The 32nd International Symposium on High Performance Liquid Phase Separations and Related Techniques, which alternates between Europe and North America, was held for the second time in Baltimore, Maryland, May 10–16, 2008. More affectionately known as HPLC 2008, the symposium is the premier scientific event for bringing together the myriad techniques related to separations in liquid and supercritical fluid media. Co-chaired by Professors Georges Guiochon of the University of Tennessee (Knoxville) and Oak Ridge National Laboratory (Oak Ridge, Tennessee) and Stephen Jacobson of Indiana University (Bloomington, Indiana), HPLC 2008 assembled just under 1200 scientists from a total of 42 countries. This number includes vendor representatives from over 67 exhibitors for the three-day instrument, software, and consumables exhibition.



Ronald E. Majors
The five-day plus event had a total of 138 oral presentations, many given during simultaneous sessions, over 460 posters in sessions with 30 themes. With an ample social event schedule, 11 vendor workshops (some with free lunch), six tutorial educational sessions, two discussion sessions, and 10 short courses, the latter held during the previous weekend, attendees had their hands full deciding how to allocate their time.
Last month, I devoted this column to the highlights of HPLC 2008 focusing on the plenary lectures, column technology, retention mechanisms including hydrophilic interaction chromatography (HILIC) and on some of the Award papers given at this important Symposium. This month, I will cover other "hot" topics including multidimensional- and comprehensive-liquid chromatography, temperature studies, application highlights, and detection techniques.
Multidimensional and Comprehensive Liquid-Phase Separations
As samples become more complex, even with the best and most efficient columns available, the peak capacity of a single column is no longer sufficient to resolve all the peaks to baseline. Peak capacities of 1000 can be achieved with a single column in a very long analysis time but to handle samples with thousands and tens of thousands of components, multiple columns with different separation mechanisms are needed. The total peak capacity of a multidimensional system is equivalent to the peak capacity of the initial column times the peak capacity of the second column. This statement is true only if the two chromatographic modes are orthogonal — totally different mechanisms of separation.
Multidimensional chromatography has been around for many years but has only achieved minimal success. With high-speed switching valves controlled by computerized systems, techniques such as heart cutting and flow diversion to eliminate uninteresting peaks are becoming more commonplace. Comprehensive liquid chromatography (LC), termed LC×LC, is a technique that also is attracting interest. In this approach, every fraction from the first dimension is switched onto a second dimension to be further separated and identified. This switching is accomplished without shutting down the flow of the first dimension column by storing the effluent from column one in a loop or packed bed and switching that storage device onto the second column once it has finished its analysis. The implication here is that the second dimension must be extremely fast in the time scale of the storage of entire effluent from column one. Readers who are interested in more details can consult a very recent review article by Schoenmakers (1).
At least four oral sessions, a tutorial, and a discussion session were devoted to comprehensive and multidimensional techniques. The one-hour tutorial session entitled "Comprehensive 2D LC: The Good, the Bad and the Ugly" was instructed by Prof. Peter Carr, University of Minnesota (Minneapolis–St. Paul, Minnesota). In the tutorial, Dr. Carr provided an historical perspective on the development of 2D LC, giving credit to the 1990 publication of Prof. Jim Jorgenson of the University of North Carolina (Chapel Hill, North Carolina) and his student Michelle Bushey, now a professor at Trinity University (San Antonio, Texas) for coining the term "comprehensive LC×LC" and demonstrating the first successful LC×LC application. The technique is recommended for complex mixtures and usually is considered a slow technique compared to 1D gradient elution. The primary (first dimension) separation column usually is run at a very low flow rate with gradient elution, and the separation in this dimension usually takes a long time, at least 30 min (sometimes longer). Microbore columns with usual flow rates in the tens of microliters per minute are suggested. In the current approach, a 10-port valve with two holding loops is used to capture the entire effluent from the primary column. The secondary (second dimension) column is usually a column designed for fast separations such as a monolith-, short-packed-, or superficially porous particle (SPP) column. As suggested, the two modes of separation should be as different as possible, truly orthogonal. Such separation orthogonality rarely exists but by adjustment of the stationary phases and the mobile phases, one can come closer to this condition.
High peak capacity is the outcome of the 2D experiment. With a single column, until now, the world record for peak capacity for a single column was 1500 peaks achieved by Richard D. Smith of Battelle Northwest National Laboratory (Richland, Washington), but the gradient separation time was 33 h and the throughput 220 samples/year. Using 2D, one can achieve high peak capacities much faster compared to 1D provided the following requirements are met:
- True orthogonality.
- Peaks cover the entire separation "space."
- The separation gained in one dimension must not be lost by separation in the second dimension.
Some of the practical issues that have to be addressed during the experiment are the numbers of samples taken from the first dimension column at the eight sigma base width of the peak (which should be at least four), solvent miscibility or compatibility, peak capture time and volume, and how to deal with the dilution that occurs. To address the latter problem, ideally one must refocus the sample after the first dimension so that one achieves a "plug" injection into the second dimension. Some of the problems to be faced in the future are: quantitation with 2D LC; long-term stability on both separation conditions; temporal alignment of peaks; improved speed in second dimension; interfacing with mass spectrometry (MS); different modes of investigation; how to deal with the 2D LC "space"; the need for data analysis software; and how to handle more than two dimensions. The big questions that came out of the tutorial and the following discussion session: what is the killer application and is 2D LC a "niche"technique or the future of LC?
A discussion session was devoted to 2D LC also led by Prof. Carr. The session had noted researchers who have made theoretical and practical contributions to advancing the status of this technique. In his opening remarks, Carr emphasized the necessity of having the second dimension a "high speed" determination that, at least until now, has limited the versatility of the 2D high performance liquid chromatography (HPLC) concept. Paola Dugo, University of Messina, Italy, indicated the most positive aspect of 2D HPLC is the enhanced resolving power, but lamented the lack of commercial support up to now. The next day, during a 2D HPLC session, Dr. Dugo gave a very practical talk on applications of the technique (see the following text).
Considered an academic toy by Andrew Shalliker of the University of Western Sydney, Australia, the current status of multidimensional techniques in industry is tied to productivity arguments. In general, the technique as practiced is too slow. Current work in multidimensional is directed towards improving the speed; Dr. Shalliker feels that particle columns will be old technology and that monolith columns will take over. However, in the long term (by 2030), both will play a minor role, as separations in space will dominate with benchtop LCx × LCx-MRI machines dominating the separations laboratory, much like LC–MS-MS is today. Space-coupled separations will yield peak capacities of 5000 or more in 0.5 h. Instruments will be low cost and MRI will read slabs or plates in 0.5-h scans and 40 proteomics samples per day will be run.
Coming back to earth, Mark Shure, Rohm and Haas (Philadelphia, Pennsylvania) felt that 2D was being held back because those "killer" applications are needed for workers to devote time and expense to this technology. Few chromatographers have tried to work in a 2D environment, although instrumentation is becoming commercially available to perform such experiments. Although fast MS detection is not essential, it will be needed to cope with the complex samples that will be encountered. The key to successful 2D will be a really fast, decent resolution second-dimension system in which the sampling of first dimension will be demanding. Most likely, 2D LC implemented with parallel LC will be pursued. Shure feels that peak capacity arguments will fall short, as many bioderived compounds are far more complex than 2D can solve.
One of the experts, Peter Schoenmakers of the University of Amsterdam, The Netherlands, indicated that trying to compare 1D LC and 2D LC was as difficult as comparing Belgian chocolate and Belgian beer. Not everything one wants is yet possible. One serious problem today is that all the method development is "trial and error" and, until this approach is solved, the 2D technique will not take off. Stated another way by one of the experts, until equipment suppliers have reliable software available off-the-shelf to handle 2D applications, the technique will lag behind.
Here are some of the areas that the experts predicted that would be addressed first:
- Polymers, surfactants, industrial chemicals (with molecular weights greater than 600).
- Proteomics (some success here but 2D LC is still not a replacement for isoelectric focusing [IEF] or sodium dodecyl sulfate polyacrylamide gel electrophoresis [SDS PAGE] yet).
- Metabolomics — this might be the key application that makes 2D LC a standard exploratory technique; perhaps more than two dimensions will be needed.
- 2D LC in process analytical technology (already some work in this direction).
Dr. Dugo gave the most practical lecture of the week on 2D LC separations by showing applications applied to real samples in the area of food chemistry such as essential oils, wine, fruit juices, and vegetable oil. Using an orthogonal combination of a cyano (normal phase) column in the first dimension and a C18 (reversed phase) column in the second dimension, she was able to profile carotenes and xantophylls in citrus fruits when combined with MS for identification. Sometimes C30 columns were required to resolve more complex samples like orange essential oils. Even 2D reversed-phase LC was used by choosing quite different hydrophobic stationary phases or different solvent combinations. Polyphenols, flavanoids and anthocyanins in wine were characterized using a 250 mm × 1.0 mm, 5-μm phenyl primary column run at very low flow rates and a 30 mm × 4.6 mm, 2.7-μm Ascentis Express C18 (Supelco, Bellefonte, Pennsylvania) or 25 mm × 4.0 mm Chromalith C18 Monolith (Merck KGaA, Darmstadt, Germany) column as the secondary column. The latter two columns were used at very high flow rates to achieve high-speed separations.
The next lecture in the same 2D session by Pavel Jandera of the University of Pardubice (Pardubice, Czech Republic) also showed some practical examples of the separation of mixtures of phenolic acids and flavones. His presentation gave guidelines for the selection of suitable phase systems for LC×LC separations. He also used resolution mapping (window-diagrams) mobile phase (or gradient) optimization strategies for each dimension to ensure mobile phase compatibility and suppression of band broadening in the sample fractions to be transferred. Sometimes, instead of sampling loops, he used short enrichment columns. A study of the effects of sampling loop volumes was undertaken for the various column dimensions. In his studies, he applied 2D reversed-phase LC with polar phases such as amide, phenyl, diol, or polyethyleneglycol for the first dimension and a monolith (C8 or C18) or superficially porous particle (SPP) (C8 and C18) phase for the second dimension. Sometimes, a gradient elution was used to improve peak capacity. Other techniques employed by his research group were covered earlier in the Best Poster section of the August "Column Watch" installment (2).
For protein and peptide separations, Mike Ramsey and his group from the University of North Carolina, experts in microfluidics, have constructed a chip made out of soda-lime glass containing a cationic coating of channels. Originally this chip was made for capillary electrophoresis (CE) and capillary electrochromatography (CEC) coupled to laser-induced fluorescence (LIF) detection. The major focus was single-cell analysis. The more recent chip design allowed for 2D-separations in time coupled to electrospray ionization (ESI)-MS. They employed the chip for the analysis of single erythrocytes. The first dimension is a pressure-driven reversed-phase separation in a channel of 100 mm length 120 μm × 25 μm i.d. packed with 5-μm Waters Symmetry C18 particles (flow rate 75 μL/min) (Milford, Massachusetts). The second dimension is a CE channel. In front of the reversed-phase column, there is a trapping column to enrich the sample. The chip is hooked up to a Nano-Acquity instrument where pressures up to 4000 psi are possible but Ramsey claims that it is safer to run the chip at 1000 psi only. Peaks eluted from the reversed-phase separation are separated by CE every 2 s.
They claim that peak capacities of 2000 should be possible with analysis times of less than 10 min. Current peak capacity achieved with the 2D chip is 230. Problems that they encounter are extremely narrow peak widths that are difficult to detect by the MS (MS-MS cycle time) and the interfacing to the Nano Acquity instrument.
In a second approach, they uncoupled the two dimensions (CE on chip, reversed phase on-column) and achieved a total peak capacity of 1700 for an E.coli digest.
Necessary improvements for on-chip integration:
- Higher performance reversed-phase separation on chip.
- Coupling of chip to LC.
- Improvement of ESI performance (bad spray).
- Faster MS.
Peter Carr also gave a lecture, in which he introduced a 2D LC system that allowed his laboratory to achieve a peak capacity of 2000 in 30 min. He combines two reversed-phase separations where the second dimension is conducted at high temperature and at a very high speed with a "large dimension" column. Sampling is achieved with two parallel loops, and it is crucial to adapt the sampling rate such that four sampling events occur across a single peak to achieve maximum peak capacity. Therefore, second dimension runs are performed in 20 s. High-temperature separations for the second dimension allow, on one side, to perform the separation fast, even with standard equipment, and also provide different selectivity compared to the ambient first-dimension separation. Fast separation at high temperature was achieved by reducing the gradient mixing volume by 95%. In addition, a second-dimension separation with a 3-s gradient on an SPP phase was demonstrated at a flow rate of 4 mL/min.
Temperature Studies in HPLC
The role of temperature has been on the list of topics for the last several years driven by the need to reduce column back pressure to more reasonable values, to increase the efficiency of separation, and by the introduction of column materials and phases that can take higher temperatures up to 200 °C.
M.J. Hayward of Lundbeck Research (Paramus, New Jersey) discussed the interaction of mobile phase gradients with temperature. Temperature can be a controlling factor in mass transfer. Thus, increasing the temperature can increase the overall speed of an analysis while maintaining resolution. As the temperature is increased, the van Deemter curve flattens, permitting higher linear velocities without prejudicing efficiencies.
J. Anspach of Phenomenex (Torrance, California) compared the van Deemter behaviors of several small particle diameter phases as a function of temperature. For most, Hmin increases as the column temperature increases, due to increases in the A-term, although the C-term becomes flatter. Some particles showed a degradation of the entire van Deemter curve above 50 °C. Higher temperatures reduce retention times, but for smaller diameter columns, the retention factor (k) must be increased to greater than 5 to compensate for extracolumn broadening.
A vendor seminar by Thermo Fisher Scientific (Waltham, Massachusetts) described the advantages that they see for high-temperature HPLC, including lower backpressures, faster throughput, faster method development, higher peak capacities, and faster analysis times. As others have shown above, for most applications, the van Deemter curve becomes flatter. One important factor to consider is that the dielectric constant is significantly temperature dependent. The example cited indicated that the dielectric constant for water at 150 °C is the same for 50:50 methanol–water at 25 °C. The implication is that less organic solvent is required to perform separations. However, the use of high temperatures, especially those that approach the boiling point of the mobile phase components, requires significant pressure reduction before the detector or a detector capable of resisting high pressure when followed by a restrictor. In either case, the detector cannot be temperature sensitive. A mobile phase preheater is also essential. Column limitations have to be taken into account since most C18 columns decompose at around 200 °C or lower. Polymeric, graphitized carbon, and some hybrid phases can take the heat. Ovens capable of high temperature operation to 200 °C are available but care must be exercised in performing rapid temperature programming because repeatability can be affected with fast ramps. Also, when using columns at high temperature, make sure that they do not have frits encased in PEEK that can liquefy at high temperature.
Selected Applications Highlights
Protein and peptide separations and proteomics again dominated liquid-phase applications. This year, the discovery of biomarkers, both large and small molecules, for disease diagnosis was a popular topic. Pharmaceutical companies are looking for tools that will be able to measure and predict the efficacy of candidate drugs in shorter times and with less expensive clinical trials. Disease-specific biomakers can speed up the process of drug discovery. For protein-based biomarkers, the use of multidimensional LC coupled with MS and MS-MS is still commanding attention. With even more techniques now available for the removal of high-abundance proteins from human plasma, trace levels of proteins are more readily detectable, although some presenters noted that sometimes small concentrations of desired proteins can tag along with the high-abundant proteins that are removed.
As usual, separations of pharmaceuticals (bulk drugs, tablets, and other formulations) including impurity profiling had a strong showing, with presentations from most of the major pharmaceutical companies. Furthermore, the analysis of drugs, drug metabolites, and endogenous compounds in biological samples and clinical chemistry continued to have a large following. Many poster presenters were involved in discussions with conferees about their experimental approaches to solve these problems.
Although few lectures were noted, the number of poster papers using CE and related techniques was quite strong again this year. This technique, which has slowed commercially, has been used in solving many problems in the life sciences, environmental science, metal analysis, and many other complex problems. The use of nonaqueous CE for chiral compounds and other pharmaceuticals continues to grow.
The papers for the separation of biomacromolecules (especially proteins) was scattered throughout the oral and poster presentations, many using mass spectral detection. Uwe Neue of Waters presented a lecture on using sub-2-μm particles with a larger pore size (285 Å) for use in the separation of biomolecules. The small particles allow fast diffusion due to the reduced diffusion distance. A short chain C4 reversed phase was found to provide good retention in gradient elution, although as expected, retention was shorter than for a C18 bonded phase. Reversed-phase chromatography of macromolecules shows an "on–off" mechanism, in which retention is a steep function of the molecular weight where the slope depends upon the log of the retention factor versus solvent composition. This factor permits a simple estimation of the HETP (H) for a protein by first measuring its peak width during the gradient and then the retention time when the protein is unretained. Using equation 1, the H value can be estimated.

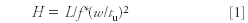
Where H = HETP, w is the peak width at half height (f = 5.55) or with tangents at the base (f = 16) measured under gradient conditions and tu is the retention time of the protein under conditions where it is unretained.
Using this approach, Neue found that for ovalbumin, the 1.7-μm wide pore particle gave better efficiency than a 3.5-μm wide pore particle. He also investigated the effect of pore size comparing 300 Å versus 130 Å. The larger pore size gave narrower peaks. High temperatures (up to 90 °C) also gave earlier elution, narrower peaks, and better peak shape. The investigation of ion-exchange moieties attached to the wide pore sub-2-μm particle showed that retention is based upon the charge of the analyte with multiple charged analytes showing stronger retention than singly charged analytes.
Caterina Temporini, University of Pavia (Pavia, Italy) introduced an integrated, on-line separation system that comprised a trypsin-based minidisk, a TiO2 trap (or in parallel a C18 trapping column), and a C18 separation column. The sample is first digested in the trypsin reactor then the flow is split and directed to a TiO2 trap or a 10 mm × 2.0 mm C18 trapping column and finally separated on a 100 mm × 1 mm C18 separation column. The focus of their study was the phosphoproteome of amniotic fluid in order to detect different degrees of phosphorylation related to fetal growth. IGF BP1, for example, has been shown to be a biomarker for birth defects. With their system, they could demonstrate that for comprehensive phosphopeptide enrichment, the desorption volume and the flow rate for the TiO2 trap is critical. While for monophosphorylated peptides, only two column volumes of high-pH solution was necessary, highly phosphorylated species required four volumes. Lower flow rates are additionally required for the desorption of highly phosphorylated phosphopeptides. With their system, they were able to identify 460 proteins with a significant percentage of phosphoproteins. Their ultimate goal is to automate the system completely but this has not been achieved.
Milos Novotny of Indiana University (Bloomington, Indiana) presented two different approaches for the analysis of glycoproteins: Glycomics, which means that the glycan residues are chopped off from the proteins and their sequence, quantity and branching pattern is analyzed; and glycoproteomics, in which glycopeptides are selectively enriched and analyzed. The first approach is interesting in terms of disease prognosis and diagnosis, where aberrant glycosylation patterns can indicate certain diseases such as rheumatoid arthritis, prion disease, cancers, and congenital disorders. Glycan chains from healthy versus diseased individuals are labeled differentially by permethylation (on-line) after selective enrichment of the glycopeptides and release of the sugar chains. Analysis is performed by matrix-assisted laser desorption ionization (MALDI)-time of flight (TOF). With this approach, it was possible to elucidate different glycosylation profiles in patients with prostate cancer versus healthy individuals.
Detection Techniques
By a perusal of the abstract, I tabulated the detection principles (Table I) that were used in the various presentations at HPLC 2008. Not every abstract indicated the detector that was used so only those that provided this information was counted. The category assignments were based on the main emphasis of a particular scientific paper as well as separation and detection techniques used. Mass spectrometry clearly dominates the detection category with UV detection a distant second. If one adds up the use of MS in chromatography and electrophoretic techniques, one half of the papers presented at HPLC 2008 used this detection technique. Compared to last year, there was even more growth in the tandem MS category where more definitive assignment of molecular structural information and greater sensitivity are achieved.

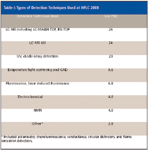
Table I: Types of Detection Techniques Used at HPLC 2008
As can be seen in Table I, LC–MS and LC–MS-MS were again the dominant detection methods used in the oral and poster presentations at HPLC 2008. When combined with the various electrophoretic techniques that were combined with MS detection, almost 50% of all papers that provided information on detection techniques used some form of MS. After MS, as might be expected, UV detection, especially diode-array detection, was the second favored detection technique, mostly in application examples.
The use of evaporative light scattering detection (ELSD) and charged aerosol detection (CAD) has been gaining in popularity. Both detectors are universal, can be used with gradients, and are more sensitive than refractive index for a variety of applications. CAD, based upon corona discharge, appears to be winning out, as poster papers on this detection method were double those of ELSD. Perhaps its higher sensitivity (up to an order of magnitude) is the reason for its popularity.
One of the more unique detection schemes was reported by Richard Zare of Stanford University (Stanford, California). He reported on the construction of a new ultraviolet thermal lensing detector based upon pulsed UV laser excitation (266 nm). So far, especially in nonpolar solvents, trace analysis is superior compared with UV absorbance. However, in water mobile phases, it is worse than UV (factor of 44) due to high signal background. The detector was used to quantitate amino acids after capillary electrophoresis separation. A total of 16 amino acids could be detected easily. The output signal had a complex dependence upon intensity due to one-photon absorption, two-photon absorption, and photon destruction of the amino acid molecules in the focus of the laser beam. Significant interference with two-photon absorption of water and the formation of multicolor centers in the fused silica flow cell caused significant interference. He demonstrated that this interference could be decreased significantly by selecting the appropriate focus and by modification of the excitation detection geometry. At the end, he claimed that as soon as further UV laser sources become available, this detector would be the method of choice for many analytes.
MS is the fastest growing detection method in HPLC. However, mass spectra generated by most ionization methods do not allow one to search a database for specific compounds because spectra are not readily available as in gas chromatography (GC) with electron ionization (EI) MS. A combined group from Agilent Technologies, (Waldbronn, Germany) and Universita di Urbino, "Carlo Bo" (Urbino, Italy) interfaced nanoHPLC to EI MS. Older techniques such as the moving belt interface and the particle beam interface never proved to be adequate and the small internal diameter columns and solvent delivery systems that were available during the early days were not as reliable as they are today. The presentation by Gerard Rozing of Agilent used packed porous particle 75-μm i.d. columns and prototype 50-μm i.d. monoliths and a nanoflow pump at flow rates of 50–500 nL/min to a heated interface to an Agilent 5975 Inert Mass Selective detector. The authors were able to demonstrate the feasibility of this combined system and assessed the ability of the system to withstand various matrix and ion suppression effects, signal-to-noise ratio versus flow rate, as well as application to real samples. With the system, matrix effects were minimal when protein precipitation, methyl tert-butyl ether (MTBE) liquid–liquid extraction, and solid-phase extraction (SPE) were investigated. Free fatty acids, high boiling organochloropesticides, and high boiling hydrocarbons provided good spectra and good sensitivity. Advantages of the technique are: it offers a cheap alternative for LC–MS, no chemical ionization and adduct formation, compatibility with nonvolatile buffers, and absence of matrix effects.
In the same LC–MS session, Prof. Barry Karger of the Barnett Institute, Northeastern University (Boston, Massachusetts) provided updated information on his interfacing of ultranarrow bore (<20-nm i.d.) LC columns to an electrospray MS system. Using porous layer open tubular (PLOT) technology, he coats polystyrene–divinylbenzene (PS-DVB) and hydrophilic interaction chromatograpy (HILIC) phases on the inside of the open tubular column. He is now investigating larger pore organic monoliths because of memory effects and with reversed-phase monolith phases has noted higher recoveries. For a 20-μm i.d. column, he was able to inject and measure glycopeptides at the 20-fmol level. The laboratory has fabricated an on-line 2D column LC–MS-MS system using a triphasic column (reversed phase–strong cation exchange–reversed phase) in the first dimension and the PS-DVB PLOT (10-μm column i.d. and 1.5-μm thick polymer layer) as the second dimension. With this system, which provides desalting and preconcentration, they have been able to identify almost 2000 proteins with the equivalent injection of total protein from ;1000 cells. With the use of a HILIC phase that uses higher organic solvent percentages than reversed-phase chromatography, improvements in ionization efficiency and a decrease in ion suppression were noted, allowing a lower injection volume.
HPLC 2009 is Next
The next major symposium in this series, the 33rd International Symposium on High Performance Liquid Phase Separations and Related Techniques (HPLC 2009), moves back to Europe and will be held for the first time in Dresden, Germany, June 28–July 2, 2009. The chairman of this upcoming event will be Prof. Christian Huber of the Saarland University (Saarland, Germany). For more information consult the official website at http://www.hplc2009.com/.
Acknowledgment
I would like to acknowledge the contributions my Agilent colleagues: summaries from Martin Vollmer and Gerard Rozing of Waldbronn, Germany, K.M Robotti from Agilent Labs in Santa Clara, California, and detailed notes from Maureen Joseph of Wilmington, Delaware. I also would like to give a special thanks to Prof. Carol Collins of State University of Campinas (Sao Paulo, Brazil) for her excellent summary of many of the columns' talks that I was unable to attend.
Ronald E. Majors
"Column Watch" Editor Ronald E. Majors is business development manager, Consumables and Accessories Business Unit, Agilent Technologies, Wilmington, Delaware, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Sample Prep Perspectives,"LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgc-mag.com.
References
(1) P. Schoenmakers, LCGC 26(7), 600–608(2008).
(2) R. Majors, LCGC 26(8), 676–691 (2008).
















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.




