Higher Order Mass Spectrometry Techniques Applied to Biopharmaceuticals
Special Issues
An outline of the basic principles of MS techniques used to investigate higher order structural features of biopharmaceuticals, as well as some insights into applications relevant to the pharmaceutical industry.
The recent trends in mass spectrometric techniques-including native mass spectrometry (MS), ion mobility spectrometry (IMS), hydrogen&ndashdeuterium exchange MS (HDX MS), and chemical cross-linking MS (CXMS)-used to elucidate higher-order structures of protein complexes and the practical implementations of these methods are discussed.
Since its invention in the early 20th century (1), mass spectrometry (MS) has been used to discover new chemical elements and their isotopes (2), explore martian soil for organic matter (3), and study biological processes by profiling proteomes or metabolomes (4,5). Biopolymers, and especially proteins, are the subject of intensive investigation. They are characterized by different levels of structural organization, ranging from the primary structure represented by the amino acid sequence, over secondary structural elements such as α-helices and β-sheets, and the three-dimensional (3D) orientation of the polypeptide chain, and finally to the assembly of subunits into protein complexes.
Analysis of protein structure using MS was first possible in the mid-1980s, when the soft ionization techniques of electrospray ionization (ESI) (6) and matrix-assisted laser desorption–ionization (MALDI) (7) were introduced. Implementation of MS for protein analysis initially focused on large-scale identification (8) and the determination or confirmation of primary structure (9), whereas newer technologies have laid the ground work for the study of tertiary and even higher order structures of protein molecules and complexes.
The analysis of biopharmaceuticals (therapeutic proteins developed for disease treatment) requires analytical techniques that are able to elucidate the various structural levels to ensure their efficacy and safety in patients. Several MS techniques are indispensable in the toolbox of physicochemical characterization methods available for the analysis of therapeutic proteins (10). Some of the methods used in the elucidation of higher order structural elements of proteins-including native MS, ion mobility MS, hydrogen–deuterium exchange MS, and chemical cross-lining MS-are discussed in this article.
Native Mass Spectrometry
Experiments using electrospray ionization mass spectrometry (ESI–MS) for the analysis of intact proteins were first performed by J.B. Fenn’s group (6). The study used strongly denaturing conditions created by using 50–90% organic solvent containing acetic acid or trifluoroacetic acid and was beneficial for the detection of multiple-charge protein ions with a quadrupole mass spectrometer with an upper nominal mass limit of m/z 1500. However, it was soon discovered that the charge-state distribution of electrosprayed proteins was significantly influenced by the protein structure prevalent under nondenaturing or denaturing conditions (11). This discovery then led to the implementation of native MS.
Native MS aims to maintain the 3D structure of proteins or protein complexes as much as possible during an experiment (12) by using conditions that reflect the protein’s native environment. The overall charge of a protein ion is limited by the number of ionizable functionalities that are accessible on the surface for charging, predominantly through protonation or deprotonation, leading to the observation of low-charged species in mass spectra. Weakly bound, noncovalent complexes-including proteins interacting with inhibitors, cofactors, metal ions, carbohydrates, or peptides-can be preserved during the electrospray process facilitating the study of the structure, stoichiometry, and association constant of such biomolecular complexes (13). The reduction of charge requires the use of mass spectrometers with an extended mass range such as time-of-flight (TOF) (14), or more recently orbital ion trap (15) mass analyzers to detect the low-charged protein species.
Trastuzumab (INN; trade name Herceptin) is a monoclonal antibody that interferes with the human epidermal growth factor receptor 2 (HER2) and is used to treat HER2-positive breast cancers. Figures 1a and 1c illustrate the difference between mass spectra of trastuzumab when analyzed under denaturing (Figure 1a) versus nondenaturing conditions (Figure 1c). Under denaturing conditions (Figure 1a), charge states from 33+ to 60+ are detected in an m/z range of 2200–4500, whereas nondenaturing conditions (Figure 1c) yield charge states of 22+ to 28+ at m/z 5200–6700. Deconvolution of both mass spectra gives equivalent masses for the uncharged species with masses in the range of 147–148 kDa and also reveals several different protein species that represent the different glycoforms of the monoclonal antibody. Such analysis can therefore readily reveal the glycosylation pattern of the protein, a highly important quality parameter of recombinant biopharmaceuticals.


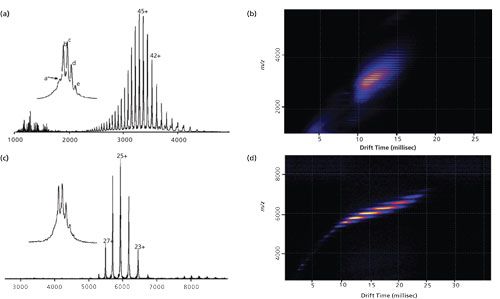
Figure 1: MS and IMS analysis of intact trastuzumab. (a) and (c): Intact MS analysis of trastuzumab. ESI-TOF mass spectra of trastuzumab in denaturing (a) or native (c) conditions. The inserts shows an extended view of the 44+ (a) and 25+ (c) charge states with resolution of the different glycoforms: (a) 147 917.1 ± 1.1 Da (G0/G0F), (b) 148 061.7 ± 0.8 Da (G0F/G0F), (c) 148 222.4 ± 0.9 Da (G0F/G1F), (d) 148 383.8 ± 0.8 Da (G1F/G1F), and (e) 148 544.3 ± 1.0 Da (G1F/G2F). (b) and (d): IMS analysis of trastuzumab. Ion mobility mobilograms of trastuzumab in denaturing (b) or native (d) conditions. IMS data obtained in native conditions (d) reveal small amounts of dimeric mAb. Adapted and reproduced with permission from reference 16, ©American Chemical Society.
Native MS has been shown to be a highly efficient tool for determining binding stoichiometry of a monoclonal antibody with its antigen. Humanized murine monoclonal antibody (hzmAb), directed against the junctional adhesion molecule A (JAM-A) to have antiproliferative and antitumoral properties, was titrated with its antigen and then analyzed using native MS to reveal noncovalent complex stoichiometries (17). Three species were detected when equimolar amounts of antibody and its target antigen were incubated including free antibody, 1:1, and 1:2 antibody&ndashantigen complex. Two molar equivalents of antigen led to an almost quantitative formation of the 1:2 complex, while eightfold molar excess yielded a 1:4 complex with a small portion of 1:3 complex.
Ion Mobility Spectrometry
Developed in the 1960s, ion mobility spectrometry (IMS) enables the generation of size- and conformation-dependent information that is not possible using MS alone. When coupled to MS this technique has the potential to separate isomers, isobars, and conformers; significantly reduce chemical background; and detect aggregates of biopharmaceuticals. IMS separation of ions is possible using differing separation powers, analyte detection, and hyphenation to mass spectrometry (18), including drift-time ion mobility spectrometry (DTIMS); aspiration ion mobility spectrometry (AIMS); field-asymmetric waveform ion mobility spectrometry (FAIMS); and traveling-wave ion mobility spectrometry (TWIMS). DTIMS and TWIMS are the two principles most often used in commercial instruments. In a DTIMS device ions are moved through a uniform, linear-field drift tube filled with a so-called “buffer gas” through a small, uniform electric field. The moving ions are attenuated by collisions with the buffer gas depending on their overall charge and collision cross-section (18). Ions with multiple charges and a small cross-sectional area move faster through the drift cell than low-charge ions with large collisional cross-sections. TWIMS is a type of IMS that utilizes traveling waves created by a series of ring-shaped electrodes that split the structure of the drift cell into a series of segments (Figure 2). Instead of a uniform linear field, a high field is applied to one segment of the cell that is subsequently swept through the cell in the direction of ion migration. Consequently, movement and separation of ions in the mobility cell is accomplished by means of pulses of an electric field passing through.

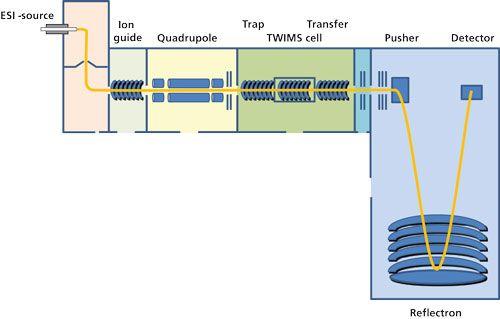
Figure 2: Schematic diagram of a quadrupole-traveling wave ion mobility&ndashtime of flight instrument.
An example for the application of TWIMS to the analysis of monoclonal antibodies is illustrated in Figures 1b and 1d. IMS analysis of trastuzumab under denaturing conditions reveals a large number of highly charged species clustering at drift times between 10 and 15 ms, while native conditions clearly distinguish between the different, low-charged species in a drift time range of 7–25 ms. This contrast in drift behavior is advantageous for the analysis of more complex mixtures of biopharmaceuticals, particularly when looking at sequence variants or other post-translational modifications such as oxidation or pyroglutamate formation.
Another practical example of IMS characterization of biopharmaceuticals (less directed towards higher order elucidation) is outlined in Figure 3. Here, a reduced mouse monoclonal antibody (IgG1, κ) sample comprising of both heavy and light chains was introduced into an ESI-quadrupole-IMS-TOF system (19). The two-dimensional (2D) ion mobilogram-mass spectrum depicted in Figure 3a clearly shows that light and heavy chains can be readily separated as different species without any other upfront separation technique. Multiple charged species related to the light and heavy chains were differentiated using mass spectra extracted from the encircled areas in Figure 3a without mutually interfering signals. Extracted mass spectra (Figures 3b and 3d) were deconvoluted using a maximum entropy algorithm, yielding spectra of uncharged species, as illustrated in Figures 3c and 3e. As expected, the light chain is detected as a single species, while the spectrum of the heavy chain reveals at least three glycoforms, characterized by different galactose content in the attached N-glycan (162 Da mass difference). This example nicely demonstrates the benefits of an additional dimension of separation, although an upfront separation method such as high performance liquid chromatography (HPLC) or capillary electrophoresis (CE) may be necessary--especially for the quantitative analysis of trace amounts of impurities that is often indispensable for quality control.

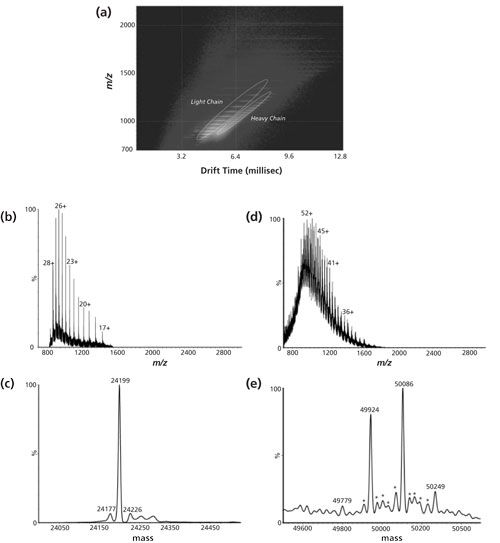
Figure 3: On-line LC-MS analysis of a completely reduced IgG1 antibody using ion mobility-TOF mode. (a) Two-dimensional plot of ion drift time versus m/z for the reduced IgG1 obtained using the ion mobility separation (7.5 V pulse). (b) Combined raw mass spectrum of the light chain. (c) Deconvoluted mass spectrum of the light chain. (d) Combined raw mass spectrum of the heavy chain. (e) Deconvoluted mass spectrum of the heavy chain. Adapted and reproduced with permission from reference 19, ©John Wiley and Sons.
Hydrogen&ndashDeuterium Exchange Mass Spectrometry
The native state of proteins is generally characterized by a tightly folded, compact structure that exposes a well-defined surface to its environment. Denaturation under conditions such as high temperature, extreme pH, adsorption to surfaces, or dissolution in organic solvent results in the protein unfolding and forming a significantly different surface. Similarly, interactions of a protein with other molecules such as small drugs, nucleic acids, or other proteins can lead to a significant change in surface properties.
Denaturing and complex formation can also have a profound influence on the exchangeability of protons at the surface of a protein molecule-the acidic protons of the carboxyl groups or acidic side chains of aspartate and glutamate are normally the most easily and rapidly exchanged while the amide protons of the protein backbone are much less prone to exchange. Exchange can be monitored by dissolution of a protein in heavy water (D2O), which leads to a hydrogen exchange by deuterium in a few minutes to hours. In proteins, the exchange rates for the different hydrogen atoms strongly depend on accessibility and therefore on protein conformation or association into higher order structures. The substitution of exchangeable hydrogen atoms with deuterium atoms forms the basis of hydrogen–deuterium exchange mass spectrometry (HDX-MS) (20).
A schematic workflow of HDX-MS is depicted in Figure 4. In brief, a protein with exchangeable hydrogens is dissolved for different periods of time at ambient or elevated incubation temperature (20–40 °C) in deuterated water (buffered to pH around 7). Depending on exchangeability, hydrogen atoms are replaced by deuterium atoms during the incubation time, before the exchange is quenched upon acidification and cooling to 0 °C. Proteins are then digested under quenching conditions, and the resulting peptides are separated by low-temperature HPLC, and finally analyzed by tandem mass spectrometry (MS/MS) upon fragmentation either by collision-induced dissociation (CID) or electron-transfer dissociation (ETD). Characteristic mass shifts in the fragment ions are indicative for the presence and positions of deuterium atoms. Analysis of the kinetics of deuterium uptake yields information about the accessibility of hydrogens at different positions in the protein, which allows valuable insights into the 3D structure of proteins or protein complexes.

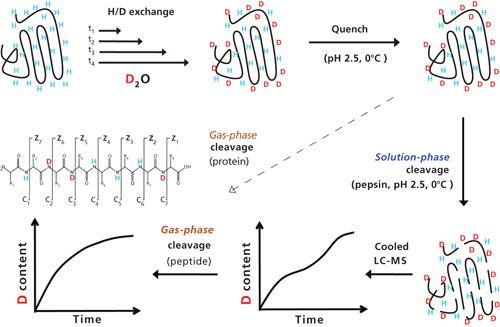
HDX-MS has been successfully used to compare 3D structures of biopharmaceuticals, which is essential to demonstrate manufacturing consistency to regulatory agencies or provide a proof of structural equivalence between an originator biopharmaceutical and its biosimilar. The advantage of this approach is that it probes the whole molecule instead of just certain substructures. The results of an interrogation of the 3D structure of interferon-β-1a, a 20-kDa cytokine used to treat multiple sclerosis (traded under the names Avonex (Biogen), Rebif (Merck Serono or Pfizer), or CinnoVex (CinnaGen) as a biosimilar, are shown in Figure 5 (22). Hydrogen exchange rates determined for five different peptic peptides effectively show the impact of different manufacturing conditions as well as post-translational modifications-modification with poly(ethylene glycol) (PEG); or oxidation at C17, M1, M36, M62, and M117-on protein structure.

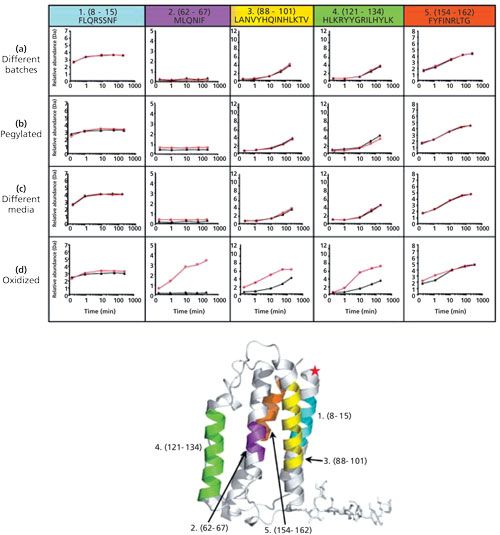
No significant alteration in hydrogen–deuterium exchange profile was observed, even though production involved different batches, using different cell media, and was subjected to N-terminal modification with PEG. A significant impact was however found for methionine or cysteine oxidation, and because the oxidized peptides incorporated more deuterium compared to the reference analogues, it could be concluded that oxidized interferon-β-1a is more solvent exposed and less hydrogen bonded.
Although this example convincingly demonstrated the applicability of HDX-MS for revealing structural differences in homogenous biopharmaceuticals, the authors also pointed out that it is not capable, at the moment, to detect conformational differences in coexisting, low-level (<10%) components of the population (22).
Chemical Cross-Linking Mass Spectrometry
Three-dimensional structures of proteins can be determined with atomic resolution by using high-resolution methods such as X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy, but the high amount of sample required for these methods (typically in the milligram range) makes them impractical for biological studies. In comparison, low-resolution structural data generated by chemical cross-linking MS (CXMS) uses much less sample amounts (in the nanogram to picogram range) to generate highly valuable data (23). Low-resolution structural information is obtained by chemically cross-linking functional groups in a protein by means of a bifunctional cross-linker, which gives information about the distance of the cross-linked functional groups in a protein molecule or a protein complex.
The most common functional groups available for cross-linking in proteins are the lysine amino groups. Sulfhydryl groups of cysteines are another possibility, but they can become involved in the 3D structure of a protein particularly when created by reduction of disulfide-bridges in the native protein. Although formaldehyde is the oldest cross-linking reagent, the most commonly utilized reagents are based on bifunctional N-hydroxy-succinimide esters, which readily react with free amino groups (and in a side reaction also with hydroxyl groups of tyrosine) to create a stable amide or imide bond upon release of N-hydroxysuccinimide. Depending on the length of the cross-linking spacer, different distances of amino acids can be probed, ranging from (almost) zero for formaldehyde to 6.4 Å for disulfosuccinimidyl tartrate, 11.4 Å for bis(sulfosuccinimidyl)suberate (10 atoms), and 16.1 Å for ethylene glycol bis(sulfosuccinimidyl succinate (14 atoms) (24).
Figure 6 shows an outline of a cross-linking experiment. After the formation of intramolecular or intermolecular cross-links, the protein or protein complex is proteolytically digested and the resulting peptides are analyzed via HPLC–MS/MS. Because the crosslink remains unaffected by the proteolysis, cross-linked amino acids are revealed through the corresponding cross-linked peptides. To more easily identify cross-linked products, isotope-labeled cross-linking reagents with 50% heavy isotope exchange can be used. Thus, cross-linked peptides are recognized by 1:1 doublets of mass signals for the light and heavy versions, which is usually achieved with the help of computer-based searching algorithms (25). The distance information obtained from the cross-linking experiment is then used to build and verify structural models for proteins or protein complexes.

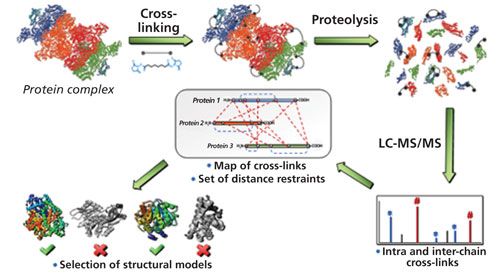
Chemical cross-linking can also be utilized to directly analyze stabilized protein complexes. For example, disuccinimidyl suberate, as well as 1,1′-(suberoyldioxyl)bisazabenzotriazole) were used as cross-linkers to stabilize the complex between the bovine prion protein and a specific antibody against it, the antibody 3E7 (26). Direct analysis of the reaction products by matrix-assisted laser desorption–ionization MS revealed both the free prion protein and the free antibody together with 1:1 and 1:2 antibody–prion protein complexes.
Conclusions
In conclusion, MS, traditionally regarded as one of the most important analytical methods for the determination of the primary structure of proteins, is increasingly contributing to the elucidation of diverse fundamental aspects of the tertiary and even quaternary structure of proteins and protein complexes. In spite of providing less spatial resolution, the major strength of MS-based investigations compared to NMR spectroscopy or X-ray crystallography lies within the comparatively low amounts of sample required for successful analysis, typically a few picograms to nanograms. Such studies are, however, only feasible with substantial support through elaborate computational algorithms and workflows, which requires significant involvement of bioinformatics into data evaluation.
Acknowledgments
The financial support by the Austrian Federal Ministry of Economy, Family, and Youth, the National Foundation of Research, Technology, and Development, and by a Start-up Grant of the State of Salzburg is gratefully acknowledged.
References
- J.J. Thomson, Proceedings of the Royal Society of London Series a-Containing Papers of a Mathematical and Physical Character89, 1–20 (1913).
- F.W. Aston, Nature105, 547–547, (1920).
- K. Biemann, Proc. Natl. Acad. Sci. U.S.A.104, 10310–10313 (2007).
- R. Aebersold and M. Mann, Nature422, 198–207 (2003).
- O. Fiehn, Plant Mol. Biol. 48, 155–171 (2002).
- C.K. Meng, M. Mann, and J.B. Fenn, Z. Phys. 10, 361–368 (1988).
- M. Karas and F. Hillenkamp, Anal. Chem. 60, 2299–2301 (1988).
- J.K. Eng, A.L. McCormack, and J.R.I. Yates, J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
- H. Nau and K. Biemann, Abstracts of Papers of the American Chemical Society 62–62 (1974).
- R.J. Falconer, D. Jackson-Matthews, and S.M. Mahler, J.Chem. Technol. Biotechnol.86, 915–922 (2011).
- J.A. Loo, H.R. Udseth, and R.D. Smith, Biomed. Environ. Mass Spectrom.17, 411–414 (1988).
- M. Przybylski and M.O. Glocker, Angew. Chem. Int. Ed.35, 806–826 (1996).
- J.A. Loo, Int. J. Mass spectrom.200, 175–186 (2000).
- M.C. Fitzgerald, I. Chernushevich, K. G. Standing, C. P. Whitman, and S. B. Kent, Proc. Natl. Acad. Sci. U.S.A.93, 6851–6856 (1996).
- S. Rosati, R.J. Rose, N.J. Thompson, E. van Duijn, E. Damoc, E. Denisov, A. Makarov, and A. J. R. Heck, Angew. Chem.-Int. Ed.51, 12992–12996 (2012).
- A. Beck, S. Sanglier-Cianferani, and A. Van Dorsselaer, Anal. Chem.84, 4637–4646 (2012).
- C. Atmanene, E. Wagner-Rousset, M. Malissard, B. Chol, A. Robert, N. Corvaia, A. Van Dorsselaer, A. Beck, and S. Sanglier-Cianferani, Anal. Chem.81, 6364–6373 (2009).
- A.B. Kanu, P. Dwivedi, M. Tam, L. Matz, and H.H. Hill, J. Mass Spectrom.43, 1–22 (2008).
- P. Olivova, W. Chen, A.B. Chakraborty, and J.C. Gebler, Rapid Commun. Mass Spectrom.22, 29–40 (2008).
- V. Katta and B.T. Chait, J. Amer. Chem. Soc.115, 6317–6321 (1993).
- K.D. Rand, M. Zehl, and T.J.D. Jorgensen, Acc. Chem. Res. 47, 3018–3027 (2014).
- D. Houde, S.A. Berkowitz, and J.R. Engen, J. Pharm. Sci.100, 2071–2086 (2011).
- A. Sinz, Mass Spectrom. Rev.25, 663–682 (2006).
- A. Sinz, J. Mass Spectrom.38, 1225–1237 (2003).
- A. Leitner, T. Walzthoeni, and R. Aebersold, Nature Protocols9, 120–137 (2014).
- C. Bich, S. Maedler, K. Chiesa, F. DeGiacomo, N. Bogliotti, and R. Zenobi, Anal. Chem. 82, 172–179 (2010).
Christian Huber was educated as an analytical chemist from 1985 to 1993 at the University of Innsbruck, Austria. Following a lecturing qualification at the University of Innsbruck in 1997, he held the chair of analytical chemistry position at Saarland University in Germany from 2002 to 2008. In 2008, he was made a professor of chemistry for biosciences at the Department of Molecular Biology of the University of Salzburg, Austria. His research interests include bioanalytical chemistry and proteome and metabolome analysis, as well as in-depth characterization of therapeutic proteins.














, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




