Evaluation of an LC Method
LCGC North America
How can we tell if an LC method is working properly?
How can we tell if a liquid chromatography method is working properly? How can a compendial method be adjusted to speed up the run time?
A liquid chromatography (LC) method from the United States Pharmacopoeia (USP) for the assay of a drug product was set up in the laboratory of one of the authors (F.O.). Although all of the details of the method cannot be shared because of its proprietary nature, the key elements follow. A 250 mm × 4.6 mm L1 column packed with 5-μm particles was used with a mobile phase of 50:50 acetonitrile–water and a flow rate of 0.6 mL/min. A refractive-index detector was used, with both the detector and column maintained at a temperature of 55 °C. System suitability requirements included a minimum resolution between the active ingredient and a related compound, a relative standard deviation (RSD) of <2% for the peak area of replicate injections of the active ingredient, and a USP tailing factor (TF) of <2. When the method was run, the resolution requirements were met; TF = 1.13 was observed, and RSD = 0.2% was determined (see Table I). It can be seen that the system suitability requirements were passed, and because the method was operated according to the monograph specifications, no further testing was required before placing the method into routine use.


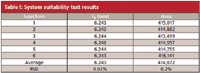
Table I: System suitability test results
Other Factors
Although the method proved suitable for use, let's examine it a little more closely to see how it fits into our expectations for a "good" isocratic method. One system suitability parameter that often is included in methods such as this is a minimum column plate number requirement. The plate number can be calculated easily from a chromatogram, such as that of Figure 1. The plate number, N, is

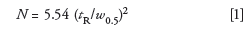
where tR is the retention time and w0.5 is the peak width at half its height, both in the same units; these values are included in the data system report generated for each chromatogram. For the last peak listed in Table I, tR = 6.243 min and w0.5 = 0.1324 min, so N = 12,300 (as usual, numbers are rounded for simplicity, so if you try to repeat the calculations presented here, you may get slightly different values).

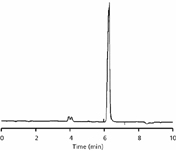
Figure 1: Chromatogram for system suitability test injection 1.
What does the calculated plate number tell us? As has been described in previous LC Troubleshooting discussions, for real samples operating under real conditions, we can estimate a reasonable plate number for any column as


where L is the column length (in millimeters) and dp is the particle diameter (in micrometers). The present column is a 250 mm × 4.6 mm column packed with 5-μm particles, so the predicted value of N ≈ 15,000. If the measured value of N is within about 20% of this predicted value, the column is in good condition. So, although the column was new, the plate number is at the low end of its expected performance range. In the present case, we'll see in a moment that the retention factor is quite small, which makes the peaks more susceptible to extracolumn band broadening because of injection problems or extracolumn volume. We suspect that this is the reason for the lower-than-expected plate number in the present example.
Retention Factor
Another parameter that can be used to evaluate the quality of an LC method is the retention factor, k. As a general rule, for the best isocratic methods, we would like to have 2 < k < 10, and if that is not possible, 1 < k < 20 usually is acceptable (see the discussion associated with Figure 1 in reference 1 for more information on this). We calculate k as


where t0 is the column dead time, usually identified as the first peak in the chromatogram or "solvent front." We can see in Figure 1 that the retention time of the first peak in the chromatogram is 4.0 min. It is a good idea to double-check that this is where the t0 peak is expected. For 4.6-mm i.d. columns we can estimate t0 from the column volume, VM:


where L is the length of the column (in millimeters) and VM is in milliliters. Therefore, in the present case VM ≈ 2.5 mL. Convert to t0 by dividing by the flow rate: t0 = 2.5 mL/0.6 mL/min = 4.2 min. The estimate should be within ≈ 10% of the observed value, so the observed t0 = 4.0 min is reasonable.
Now we can calculate k = (6.2 – 4.0)/4.0 = 0.6. This is much less than even the minimum desired k = 1. Does this mean that the method is bad or that a bad method was approved and included in the USP? Not necessarily; let's look further.
The L1 Conundrum
Let's take a look at the column that is used for the method, which USP classifies as L1. L1 columns are defined as "octadecylsilane (C18) chemically bonded to porous silica . . . micro-particles" (p. 982 in reference 2). This reflects the belief many years ago that all C18 columns are the same or very similar. Of course, today we recognize that this is not true, and it is not difficult to find two C18 columns that differ by a factor of two in the retention of any particular compound. Although it is permissible to use any L1 column with the present method, it would be much more appropriate to use the same brand of column that was used when the method was developed for the USP monograph. Fortunately, the USP website (3) contains a list of which columns are associated with monograph methods. When we consulted the database, we found that the recommended column was a 250 mm × 4.6 mm Inertsil ODS-3 column packed with 5-μm particles (GL Sciences). From data we have available (for example, Table 7.2 in reference 4), we observed that the column that was used to generate the data of Figure 1 and Table I gives k values of about 60% of the Inertsil column. If this same retention ratio is true for the analyte of interest, the k value would increase from ~0.6 to ~1.0, which would move it into a more reasonable range. This means that the lower-than-desired retention factor observed with the method could be attributed to the choice of a different L1 column than the one used for the original method, and not a poorly designed method.
Possible Method Improvements
One reason to use existing USP methods is that they do not need to be validated if the method can be set up and shown to pass the system suitability requirements. This is an obvious savings of time and money, so it is understandable that most users of USP methods are reluctant to make any changes. However, in Chapter 621 of the USP (2), there is a list of adjustments that can be made to existing methods that do not require revalidation, providing, of course, that system suitability still passes. If we consider the allowable changes and the simplicity of the present method, it is likely that we can shorten the run time without compromising the results.
First, let's consider the column size. The method designates a 250 mm × 4.6 mm column. This suggests to us that the method is quite old — 250-mm columns rarely are used with methods today. The USP allows a change in column length of ±70%, so a change to a 150-mm-long column would be a reduction of only 40% in length, well within the allowance. This adjustment would reduce the overall run time from 10 min to 6 min and the retention of the analyte from 6.2 min to 3.7 min. Because resolution changes only with the square root of the plate number (and thus square root of the column length change), the shorter column is unlikely to compromise resolution in the current method, where the analyte of interest is well resolved from the impurity.
The USP also allows adjustment of the flow rate by as much as ±50%, meaning that the 0.6-mL/min flow rate could be increased to 0.9 mL/min. A more careful reading of the actual method states that the flow rate is "about 0.6 mL per minute," so it would be acceptable to run the method at 1 mL/min with the adjustment. This would further reduce the run time from 6 min to 3.6 min and the retention time from 3.7 min to 2.3 min. Again, resolution is unlikely to be affected by the flow rate change because for 5-μm and smaller particle sizes a change of flow rate by a factor of two will result in only a small change in the plate number for most methods.
Two other minor changes are suggested by an examination of the chromatogram of Figure 1. First, the end-integration marker (vertical bar below baseline at ~7 min) appears to be a bit later than is necessary. This could be adjusted to force the peak to end closer to where the peak returns to baseline — for example, at 6.5 min. Second, the negative peak between 8 and 9 min appears to be the last peak in the chromatogram. After this peak has been eluted, there is no need to continue the run, so the run time could be reduced from 10 min to 9 min. This 10% reduction in run time would also apply to the corresponding run-time changes for a shorter column or higher flow rate, or both.
If the changes in column length or flow rate were made, care should be taken to make sure that system suitability still passed. Special attention should be paid to the minimum resolution requirements to separate the active ingredient from the related compound. Providing that these adjustments still result in acceptable method performance, the changes should be documented, along with a comparison of the method performance (for example, precision, accuracy, resolution, and tailing factor) before and after the change. This documentation will be useful if, during a regulatory inspection, the validity of the changes is questioned.
Conclusions
We observed that the LC method could be set up and would pass the system suitability requirements. Although technically this is all that needs to be demonstrated to permit use of a compendial method, it is a good idea to check some performance factors that are not required, but can be used to help assure us that the method is operating properly.
The column plate number is easy to determine, and often it can be calculated automatically by the chromatographic data system. We used an estimate of N to determine that the observed plate number is reasonable; so the column is in good condition.
We also calculated the retention factor to see if the retention time of the analyte of interest was in a range that is desired for a robust LC method. We found that, although the retention time seemed to be large, the k value was less than desired. In some methods, this would raise a red flag, but there are several factors that reduce our concern about the low k value in the present application. First, the chromatogram is very simple — there is only one peak that we are interested in, and as long as it is separated adequately from a particular related compound, there is little concern. Second, there is very little disturbance in the baseline at t0, suggesting that there is little likelihood that unretained materials will interfere with the peak of interest. Third, the repeatability of peak areas and retention times was excellent, which tells us that even though the k value is less than desired, the method performs acceptably for the present application. It also is likely that the low k value contributes to the slightly lower than expected plate number for the column. The smaller the k value, the more likely that extracolumn effects, such as plumbing and injection conditions, will cause peak broadening. Even so, the plate number is acceptable and peak tailing is minimal, so there is little reason for concern.
This method is a good example of a case where the perfect method is not necessary to get acceptable results. Any changes to the method other than the change in column length and flow rate discussed here would be likely to require some level of method revalidation. Validation is a time-consuming and expensive process, so any additional reduction in method run time would be offset by these extra costs. This is a good example of the maxim, "better is the enemy of good enough."
References
(1) J.W. Dolan, LCGC North Am. 30(9), 828–832 (2012).
(2) United States Pharmacopeia 34–National Formulary 29 (United States Pharmacopeial Convention, Rockville, MD, 2011).
(3) http://www.uspchromcolumns.com/chrom/display?cmd=index.
(4) L.R. Snyder, J.W. Dolan, D.H. Marchand, and P.W. Carr, Adv. Chromatogr. 50, 297–376 (2012).
Fatos Osmani is a senior HPLC analyst at Profarma in Tirane, Albania.


Fatos Osmani
John W. Dolan "LC Troubleshooting" Editor John Dolan has been writing "LC Troubleshooting" for LCGC for more than 25 years. One of the industry's most respected professionals, John is currently the Vice President of and a principal instructor for LC Resources, Walnut Creek, California. He is also a member of LCGC's editorial advisory board. Direct correspondence about this column via e-mail to John.Dolan@LCResources.com.


John W. Dolan










; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")




. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

