Elevated Temperatures in Liquid Chromatography, Part I: Benefits and Practical Considerations
LCGC North America
Benefits of using elevated-temperature separations and some practical considerations for implementing this approach.
Elevated-temperature separations offer significant benefits compared to those performed at ambient temperature. Here, we discuss 12 such benefits, and then describe some practical considerations for implementing elevated-temperature liquid chromatography.
In part I of this series we discuss the various advantages and practical considerations of performing elevated temperature liquid chromatography (LC) separations. In part II, we will continue our discussion of this topic, exploring some basic thermodynamic relationships, including the derivation and use of the van 't Hoff equation. Finally, in part III we will present a more detailed discussion of the van 't Hoff equation, exploring its usefulness and relevance using various examples from the literature.
Benefits of Elevated Temperature LC
Elevated-temperature separations offer many benefits, which are discussed below.
Lower Column Back Pressures and Faster Separations
Because solvent viscosity is a strong function of temperature, elevated temperatures decrease mobile-phase viscosities, which leads to lower column back pressures and faster separations. The dependence of viscosity on temperature is exponential and is given by the following equation:



where η is the viscosity, a, b, and c are coefficients that are specific to a solvent, and T is the temperature in kelvins (1). Accordingly, noticeable reductions in viscosity, and therefore back pressure, are observed in systems running even at relatively low temperatures. For example, the viscosity of water decreases from 0.890 centipoise (cP) to 0.547 cP and acetonitrile decreases from 0.369 cP to 0.284 cP at 25 °C and 50 °C, respectively (2). This advantage only increases with increasing temperature: A 20-fold improvement in analysis time can be realized when working at 150–200 °C (3–5).
"Greener" Modifiers Can Be Used
With the reduction in mobile-phase viscosity with increasing temperature, less toxic and "greener," but inherently more viscous, organic modifiers can be contemplated, such as ethanol. Separation of 18 fat- and water-soluble UV filters for cosmetic formulations was achieved using ethanol as an organic modifier under gradient conditions at 45 °C (6). Ethanol and water were also used to separate various phthalates at 35 °C (7).
Temperature Can Exceed Solvent Boiling Point
Column temperatures exceeding the normal boiling points of the typical solvents used in LC can be used in elevated-temperature LC because a relatively inexpensive back-pressure regulator can be installed at the end of the system, such as after the UV detector, to suppress boiling of the mobile phase in the column. The typical pressure range for a back-pressure regulator is 20–70 bar.
The increase in solvent boiling point with external pressure follows from the integrated Clausius-Clapeyron equation (8):

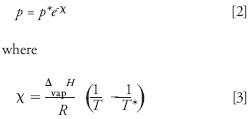
In these equations, p* is the vapor pressure of the liquid at a temperature of T*, p is the vapor pressure at the temperature T, ΔvapH is the enthalpy of vaporization of the liquid, and R is the gas constant. Values of ΔvapH for various solvents of importance in chromatography are given in Table I. Because a liquid boils when its vapor pressure exceeds the pressure acting on it, it is clear that the column pressure must be kept above the vapor pressure of the mobile phase to prevent it from boiling. A back-pressure regulator of approximately 30 bar is usually sufficient to keep typical mobile phases from boiling (9).


Table I: Enthalpy of vaporization (ÎvapH) for various solvents (2). Note that enthalpies are generally fairly constant with temperature.
Water Behaves More Like an Organic Solvent
As the temperature increases, the static permittivity of water decreases, so water behaves more and more like an organic solvent. Thus, at elevated temperatures, water is increasingly able to dissolve nonpolar species (10), and separations using more aqueous mobile phases can mimic those using more organic modifiers at lower temperatures. For neutral compounds on silica- and zirconica-based columns, an increase of 4–5 °C affects retention like a 1% increase in organic modifier (11–13). More specifically, a temperature increase of 3.75 °C has a similar elution strength change as a 1% increase in methanol (14), and a 5 °C increase in temperature was comparable to a 1% increase in acetonitrile (11). Figure 1 shows graphically how heated water can mimic the elution strengths (dielectric constants) of water–methanol or water–acetonitrile mixtures. For example, to replace a separation that uses 40:60 water–methanol (v/v) with a pure aqueous mobile phase the chromatographer would operate near 130 °C.


Figure 1
In some applications pure aqueous mobile phases have been used at temperatures exceeding 100 °C. This method has been called "superheated water chromatography," "subcritical water chromatography," or "chromatography in very hot water" (13). Figure 2 shows the separation of several steroids at various temperatures using a superheated water mobile phase (18). The separation performed at 120 °C was far from ideal. As the temperature was increased to 140 °C and then 185 °C, the analysis time was reduced tremendously: from 30 min to 18 min to 5.5 min. Notice that the degree of tailing decreased with increasing temperature, which can be attributed to a decrease in the number and effectiveness of secondary interactions.

Figure 2
The van Deemter equation sheds light on some of the benefits of elevated-temperature LC:

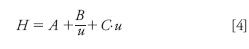
where H is the height equivalent of a theoretical plate; A is the eddy diffusion coefficient, which depends on the quality of the column packing and the size of the particles in the packed bed, but is otherwise unaffected by temperature; B is the longitudinal diffusion term; C is the mass transfer coefficient; and u is the linear velocity of the mobile phase. A simplistic breakdown of the B and C terms shows that both depend on the analyte diffusion coefficient, Dm, as follows (1):

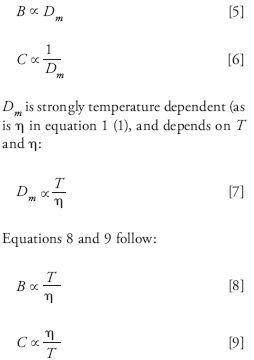
Hence, as T increases, B increases and C decreases. As a result, an increase in temperature will increase the degree to which longitudinal diffusion occurs (the B term), unfavorably raising plate heights and broadening bands. However, elevated temperatures lead to more efficient solute and mass transfer between the mobile and stationary phases, and diffusion is accelerated within the mobile phase itself (20,21), which reduces resistance to mass transfer of the analyte (the C term), and lowers plate height and is favorable. Two important consequences of these effects are that as temperature increases, the minimum in the van Deemter equation (Hmin) shifts to higher values of u (21,22) and van Deemter curves flatten (see Figure 3). Thus, elevated temperatures allow for faster separations with little or no loss of efficiency. Note that in general, Hmin stays roughly constant with increasing temperature, so efficiencies do not change significantly with changing temperature as long as the linear velocity of the separation is near the optimum.


Figure 3
An empirical demonstration that higher flow rates do not strongly affect the quality of a separation at elevated temperature (185 °C) is shown in Figure 4. At 1 mL/min, four steroids were separated in <5.5 min (19). Under identical conditions, the same separation was then performed in <1.5 min using a flow rate of 5 mL/min (18). Even at this greatly increased flow rate, the steroids were still baseline separated.

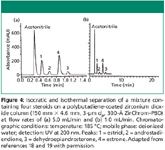
Figure 4
Other Advantages
There are a number of additional advantages when using elevated temperature separations. These are described below.
Temperature is an instrumental variable, not a chemical variable, so with proper equipment it is often easier to change the temperature of a separation than to explore a chemical change in the mobile or stationary phase.
Changes in analyte selectivity with temperature can sometimes be substantial, and reversal of elution order of certain analytes with increasing temperature may be observed (25). For example, while a decrease in retention times of analytes is usually found with increasing temperature in reversed-phase LC, McCalley (26) demonstrated an increase in retention times of various bases with increasing temperature. This increase in retention could be attributed to a temperature dependency of the pKa of the ionizable portion of the base.
Elevated-temperature LC raises the possibility of performing retention modeling over a range of temperatures — that is, predicting and optimizing the retentions of analytes with input from relatively few experiments (27–32). Because plots of ln k vs. 1/T are often linear, and because elevated-temperature LC is increasingly practiced, this advantage is becoming particularly important (33).
The option of direct extraction of thermodynamic data through van 't Hoff plots, which are plots of ln k vs. 1/T, where k is the retention factor and T is the temperature in kelvins. This information can serve both to characterize a column and to allow its comparison to others (34,35).
In general, thermostating a column at a temperature above room temperature is a very good idea to avoid the daily fluctuations in temperature that take place in most laboratories and that can change analyte retention. Analyte retention typically changes by approximately 2% for each 1 °C change, so an intraday or day-to-day change in laboratory temperature of ±5 °C or more, which can occur in many laboratories, can substantially change analyte retentions (20,36–38).
The equipment needed for elevated-temperature separations is readily available; relatively inexpensive column ovens are commercially available and provide temperature control and the necessary precolumn heating and postcolumn cooling of the mobile phase (36,39,40–42). Many high performance liquid chromatography (HPLC) and ultrahigh-pressure liquid chromatography (UHPLC) systems sold today come with column ovens.
Analyte decomposition in elevated-temperature LC is not generally an issue, because most analytes possess good thermal stability, even at relatively high temperatures, during their short dwell times in a column (3). However, in the case that a thermally labile compound may be part of a separation, or that a reaction may be possible between the mobile phase and an analyte, the questions presented by Thompson and colleagues (3) may help chromatographers decide whether elevated-temperature chromatography is desirable: Does working at elevated temperature reduce the dependability of the analytical calibration curve? Does elevated temperature chromatography induce a significant intercept in the calibration curve? Does temperature significantly diminish the sensitivity? If present, do on-column reactions distort peak shape? Does elevated-temperature chromatography introduce new peaks that interfere with the resolution and quantitation of the analyte or impurities? Pursuant to an impurity analysis, does a high-temperature mobile phase cause chemical reactions that alter the concentration or produce products that would interfere with quantitation of the impurity? If the answer to these questions is no, then elevated-temperature chromatography should be a viable option.
Practical Considerations for Elevated-Temperature LC
To realize the advantages of elevated-temperature HPLC, the mobile-phase flow rate must increase and the column oven must be equipped with an efficient, low-volume means of properly adjusting the mobile-phase temperature (4). That is, the mobile phase needs to be preheated before entering the chromatographic column to avoid a thermal mismatch between the column and the incoming mobile phase. If a thermal mismatch exists, then band broadening of the peak may result, producing less-than-optimal chromatography. This thermal mismatch should be kept to less than 5 °C (3). Many systems come equipped with column preheaters that can be thermostated independently of the column oven. Figure 5 shows a diagram of a typical elevated-temperature apparatus. As with most systems, the solvents are mixed after the pumps and before the injector. The incoming solvent is preheated. The column is thermostated at a particular temperature via a column oven, and the effluent is cooled before it enters the detector. As discussed earlier, the column is maintained under a certain pressure via a back-pressure regulator that is placed in-line after the detector and before waste.

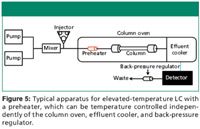
Figure 5
Figure 6 demonstrates how band broadening may occur if the mobile phase is not properly preheated. In Figure 6a the temperature of the incoming mobile phase matches the temperature of the column. When the temperatures of the mobile phase and column match, peak distortion resulting from thermal gradients is avoided. In Figure 6b the incoming mobile phase is cooler than the column, which leads to radial thermal gradients — that is, the center of the column will be cooler than the outer portion of the column. This type of thermal mismatch between the column and mobile phase will result in broader peaks and less than optimal chromatography. In Figure 6c frictional heating is present. Frictional heating becomes a problem at high pressures because of friction between the mobile phase and the stationary phase or support. Frictional heating may lead to axial (along the length of the column) and radial thermal gradients. Axial thermal gradients also result in band broadening and a loss of efficiency. Figure 6d shows that one may compensate for frictional heating by introducing the mobile phase at a reduced temperature.

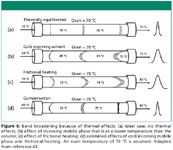
Figure 6
A chromatographic demonstration of the thermal mismatch diagrammed in Figure 6 is shown in Figure 7. Figure 7a shows an ideal chromatogram obtained by preheating the mobile phase to the temperature of the column. Figure 7b shows a chromatogram with the incoming mobile phase cooler than the column. This thermal mismatch produces two effects: the retention times of peaks 3–7 are changed when compared to the chromatogram in Figure 7a (they are longer because the temperature of the mobile phase is lower), and peaks 6 and 7 are broader and distorted, a result that is attributed to thermal gradients in the column (43,44). Figure 7c also shows a thermal mismatch between the entering mobile phase and the column, but in this case these temperatures were adjusted to produce the same retention times as in the ideal chromatogram shown in Figure 7a. However, peaks 3, 6, and 7 are still distorted, and peaks 6 and 7 are noticeably split. These less-than-desirable effects are again attributed to axial and thermal gradients in the column.

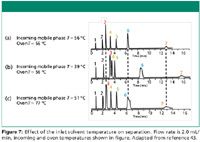
Figure 7
As a final practical consideration, one must be aware of the temperature limits of any columns that might be contemplated for use in elevated temperature chromatography. Many stationary phases or supports, especially those based on silica, degrade quickly at elevated temperatures. These effects are exacerbated at extremes of pH. However, there are also some materials based on silica that exhibit excellent stability even at temperatures above 100 °C (45).
References
(1) S. Giegold, T. Teutenberg, J. Tuerk, T. Kiffmeyer, and B. Wenclawiak, J. Sep. Sci.31(20), 3497–3502 (2008).
(2) CRC Handbook of Chemistry and Physics, D.R. Lide, Ed. (CRC Press, Boca Raton, Florida, 2004).
(3) J.D. Thompson and P.W. Carr, Anal. Chem. 74(5), 1017–1023 (2002).
(4) F.D. Antia and C. Horváth, J. Chromatogr. A 435, 1–15 (1988).
(5) T. Teutenberg, S. Wiese, P. Wagner, and J. Gmehling, J. Chromatogr. A 1216(48), 8470–8479 (2009).
(6) A. Salvador and A. Chisvert, Anal. Chim. Acta 537(1–2), 15–24 (2005).
(7) D.D. Orsi, L. Gagliardi, R. Porrà, S. Berri, P. Chimenti, A. Granese, I. Carpani, and D. Tonelli, Anal. Chim. Acta 555(2), 238–241 (2006).
(8) P. Atkins and J. De Paula, in Atkins' Physical Chemistry, (W.H. Freeman and Company, New York, New York, 2006).
(9) C.V. McNeff, B. Yan, D.R. Stoll, and R.A. Henry, J. Sep. Sci. 30(11), 1672–1685 (2007).
(10) A.M. Edge, S. Shillingford, C. Smith, R. Payne, and I.D. Wilson, J. Chromatogr. A 1132(1–2), 206–210 (2006).
(11) C.M. Hong and C. Horváth, J. Chromatogr. A 788(1–2), 51–61 (1997).
(12) J.V. Tran, P. Molander, T. Greibrokk, and E. Lundanes, J. Sep. Sci. 24(12), 930–940 (2001).
(13) S. Heinisch and J.-L. Rocca, J. Chromatogr. A 1216(4), 642–658 (2009).
(14) J. Bowermast and H.M. McNair, J. Chromatogr. Sci. 22, 165–170 (1984).
(15) C.A. Meyer, in ASME steam tables: thermodynamic and transport properties of steam: comprising tables and charts for steam and water, calculated using the 1967 IFC formulation for industrial use in conformity with the 1963 international skeleton tables, as adopted by the Sixth International Conference on the Properties of Steam, (American Society of Mechanical Engineers, New York, new York, 1983).
(16) L.G. Gagliardi, C.B. Castells, C. Ràfols, M. Rosés, and E. Bosch, J. Chem. Eng. Data 52(3), 1103–1107 (2007).
(17) G. Akerlof, J. Am. Chem. Soc. 54(11), 4125–4139 (1932).
(18) T. Teutenberg, H.J. Goetze, J. Tuerk, J. Ploeger, T.K. Kiffmeyer, K.G. Schmidt, W.g. Kohorst, T. Rohe, H.D. Jansen, and H. Weber, J. Chromatogr. A 1114(1), 89–96 (2006).
(19) T. Teutenberg, in High-Temperature Liquid Chromatography A User's Guide for Method Development, R.M. Smith, Ed. (RSC Publishing, Cambridge, UK, 2010).
(20) G. Vanhoenacker, F. David, and P. Sandra, Chromatogr. Today 3(3), 14–16 (2010).
(21) Y. Xiang, B. Yan, B. Yue, C.V. McNeff, P.W. Carr, and M.L. Lee, J. Chromatogr. A 983(1–2), 83–89 (2003).
(22) G. Vanhoenacker and P. Sandra, Anal. Bioanal. Chem. 390(1), 245–248 (2008).
(23) F.D. Antia and C. Horváth, J. Chromatogr. A 435(0), 1–15 (1988).
(24) J. Li and P.W. Carr, Anal. Chem. 69(5), 837–843 (1997).
(25) A.M. Edge, S. Shillingford, C. Smith, R. Payne, and I.D. Wilson, J. Chromatogr. A 1132(1–2), 206–210 (2006).
(26) S.M.C. Buckenmaier, D.V. McCalley, and M.R. Euerby, J. Chromatogr. A 1060(1–2), 117–126 (2004).
(27) J.W. Dolan, J. Chromatogr. A 965(1–2), 195–205 (2002).
(28) J.W. Dolan, L.R. Snyder, N.M. Djordjevic, D.W. Hill, D.L. Saunders, L. Van Heukelem, and T.J. Waeghe, J. Chromatogr. A 803(1–2), 1–31 (1998).
(29) J.W. Dolan, L.R. Snyder, D.L. Saunders, and L. Van Heukelem, J. Chromatogr. A 803(1–2), 33–50 (1998).
(30) J.W. Dolan, L.R. Snyder, N.M. Djordjevic, D.W. Hill, and T.J. Waeghe, J. Chromatogr. A 857(1–2), 1–20 (1999).
(31) J.W. Dolan, L.R. Snyder, N.M. Djordjevic, D.W. Hill, and T.J. Waeghe, J. Chromatogr. A 857(1–2), 21–39 (1999).
(32) J.W. Dolan, L.R. Snyder, R.G. Wolcott, P. Haber, T. Baczek, R. Kaliszan, and L.C. Sander, J. Chromatogr. A 857(1–2), 41–68 (1999).
(33) S. Wiese, T. Teutenberg, and T.C. Schmidt, Anal. Chem. 83(6), 2227–2233 (2011).
(34) L.A. Cole, J.G. Dorsey, and K.A. Dill, Anal. Chem. 64(13), 1324–1327 (1992).
(35) L.A. Cole and J.G. Dorsey, Anal. Chem. 64(13), 1317–1323 (1992).
(36) S. Heinisch and J.-L. Rocca, J. Chromatogr. A 1216(4), 642–658 (2009).
(37) F. Gritti and G. Guiochon, J. Chromatogr., A 1099(1–2), 1–42 (2005).
(38) J.W. Dolan, LCGC North Amer. 21(3), 262–266 (2003).
(39) R.G. Wolcott, J.W. Dolan, L.R. Snyder, S.R. Bakalyar, M.A. Arnold, and J.A. Nichols, J. Chromatogr. A 869(1–2), 211–230 (2000).
(40) T. Teutenberg, H.J. Goetze, J. Tuerk, J. Ploeger, T.K. Kiffmeyer, K.G. Schmidt, W.g. Kohorst, T. Rohe, H.D. Jansen, and H. Weber, J. Chromatogr. A 1114(1), 89–96 (2006).
(41) Selerity Technologies, Inc. http://www.selerity.com/main/main_products_hplc_9000.html.
(42) Scientific Instruments Manufacturer GmbH http://www.sim-gmbh.de/index.php?option=com_content&task=view&id=64&Itemid=502&lang=en.
(43) R.G. Wolcott, J.W. Dolan, L.R. Snyder, S.R. Bakalyar, M.A. Arnold, and J.A. Nichols, J. Chromatogr. A 869(1–2), 211–230 (2000).
(44) D. Guillarme, S. Heinisch, and J.L. Rocca, J. Chromatogr. A 1052(1–2), 39–51 (2004).
(45) T. Teutenberg, K. Hollebekkers, S. Wiese, and A. Boergers, J. Sep. Sci. 32(9), 1262–1274 (2009).
David S. Jensen and Matthew R. Linford are with the Department of Chemistry and Biochemistry at Brigham Young University in Provo, Utah. Direct correspondence to: mrlinford@chem.byu.edu.
Thorsten Teutenberg is with the Institut für Energie- und Umwelttechnik e. V. in Duisburg, Germany.
Jody Clark is with Selerity Technologies Inc., in Salt Lake City, Utah.













Characterizing Plant Polysaccharides Using Size-Exclusion Chromatography
April 4th 2025With green chemistry becoming more standardized, Leena Pitkänen of Aalto University analyzed how useful size-exclusion chromatography (SEC) and asymmetric flow field-flow fractionation (AF4) could be in characterizing plant polysaccharides.




Investigating the Protective Effects of Frankincense Oil on Wound Healing with GC–MS
April 2nd 2025Frankincense essential oil is known for its anti-inflammatory, antioxidant, and therapeutic properties. A recent study investigated the protective effects of the oil in an excision wound model in rats, focusing on oxidative stress reduction, inflammatory cytokine modulation, and caspase-3 regulation; chemical composition of the oil was analyzed using gas chromatography–mass spectrometry (GC–MS).

