The Effect of Modifier on Selectivity in Reversed-Phase High Performance Liquid Chromatography
LCGC Europe
The retention behaviour of several compounds has been compared for their selectivity using reversed-phase high performance liquid chromatography with binary water mobile phases composed of methanol, acetonitrile or tetrahydrofuran as modifiers.
Reversed-phase high performance liquid chromatography (RP-HPLC) is the most popular method to separate and analyse chemical compounds in pharmaceutical, biomedical and environmental samples.1 The choice of appropriate conditions is a challenging problem in HPLC analysis.2,3 These conditions depend on the type of the stationary phase, the qualitative and quantitative composition of the eluent, and the kinetic properties of the chromatographic system. Chromatographic optimization of the separation process is difficult to perform, particularly when the composition of the sample mixture is complicated. Such optimization problems are usually solved on the basis of the chromatographer's experience and theoretical knowledge. Computer software4–7 can be applied to assist this optimization process. However, in each stage of the optimization procedure an understanding of the chromatographic process at the molecular level is still necessary. This understanding helps when optimizing the separation process, particularly for complex sample mixtures. There have been several approaches to explain retention and selectivity changes in liquid chromatography — the most popular of these is solvophobic theory proposed by Horvath et al.8,9
Retention and selectivity can be predicted based on the semi-empirical correlation between retention factor, k, and the composition of the mobile phase.10,12 Selectivity changes should be expected when the solvent component (modifier) of the mobile phase is replaced by another in the chromatographic system, based on the different abilities to form dipolar, proton donor and proton acceptor interactions with the solute molecules. These different solvent abilities to form molecular interactions with solute molecules can be predicted by the solvent selectivity triangle.13,15
However, prediction of selectivity changes, especially for RP systems when the modifier is changed in the mobile phase, is often ambiguous. For example, if the retention of solute A is increased relative to solute B because the modifier is changed in the mobile phase, then this effect can be interpreted as the enhancement of molecular interactions of this solute in the stationary phase and/or the reduction of molecular interactions in the mobile phase. This approach does not explain selectivity changes clearly and can lead to some confusion.
Therefore, an explanation of the selectivity changes resulting from the molecular interaction of the solute in one phase only would eliminate the uncertainty. This approach assumes that selectivity changes when one modifier is replaced by another in the eluent. It can be explained by considering the molecular interactions between solute and modifier in the stationary phase (interactions in the mobile phase are neglected) and ordering of the stationary phase by the modifier.16
This hypothesis was confirmed by the data presented in reference 16 in which the gas–liquid partition constant, K, of benzene and its seven derivatives with different functional groups (acetophenone, benzaldehyde, benzonitrile, chlorobenzene, nitrobenzene, phenol and toluene) were compared based on the experimental data from reference 17. The liquid phase was composed of water and organic solvent [48.4% methanol (MeOH), 29.7% acetonitrile (ACN) or 29.7% tetrahydrofuran (THF)]; that is, with modifier concentration often applied in RP-HPLC systems.
The correlation coefficient, R, of the relationship between log KA versus log KB, where A and B denote two different gas–liquid systems, respectively, shows values greater than 0.995 for all correlated systems; for example, MeOH versus ACN, MeOH versus THF and ACN versus THF.
This means that partition selectivity in the gas–liquid system does not depend on the modifier type, or its influence on this partition is minor in the restricted range of modifier concentration in the liquid solution. Based on these results it was concluded that similar relationships could occur in an RP-HPLC system19 (i.e., selectivity changes when the organic component of the mobile phase is replaced by another). This can be explained by molecular interactions of the solute with components of the stationary phase of RP-HPLC system only.
In this approach interactions of the solute with components of the mobile phase can be omitted. The discussed approach can additionally be corroborated with regards to solvophobic properties of the mobile phase. It should be mentioned that the cohesion energy of water is 554 cal/cm3 . The organic components of the mobile phase have much lower cohesion energies (MeOH: 210 cal/cm3 , ACN; 174 cal/cm3 , THF: 98 cal/cm3 ). It is evident that water is responsible for hydrophobic expulsion of the solute molecules from the mobile phase when its concentration is relatively high.16,18
The stationary phase of the RP-HPLC system contains hydrocarbon chains, unreacted silanol groups, water and extracted modifier molecules. The composition of the first two components in this list is constant. The concentration of water (present in stationary phase mainly because of solvatation of the silanol groups and the modifier molecules) is constant in the restricted range of modifier concentration. Therefore, the fourth component (modifier) is responsible for differences of the properties of the stationary phase when the organic component is changed in the eluent.
These properties of the stationary phase vary when the mobile phase modifier is replaced by another. This means that selectivity changes of the RP-HPLC system can be explained by the molecular interactions of the solute in the stationary phase when organic component of the mobile phase is changed. This discussion was confirmed by thermodynamic considerations that lead, after some simplifications, to the equation for difference of free energy change, Δ (ΔG0) of the solute RP-LC systems with different modifiers:19


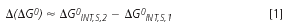
where 1 and 2 correspond to systems with modifier 1 and 2, respectively, and ΔG0INT s is the free energy change as a result of interactions of the solute molecule with surrounding components (including extracted modifier) of the stationary phase.
Change of modifier in the eluent leads to alteration of the molecular interactions in the stationary phase, which are responsible for separation selectivity.20 This means that selectivity variation can be explained, when modifier is changed in the mobile phase, by interaction of the solute molecules with components of the stationary phase, especially with molecules of organic solvent extracted into the region of non-polar ligands (hydrocarbon chains) of the stationary phase. The ordering of the stationary phase region, which depends on the modifier type, influences selectivity changes concerned with the various shapes of the molecules of the solutes to be separated. The ability of the modifier to increase the ordering of the hydrocarbon stationary phase rises in the following order: MeOH, ACN, THF.


Keynotes:
The ordering caused by THF is particularly evident because of its properties: strong sorption (Figure 1),21 and a relatively large and almost planar structure compared with the remaining modifiers (MeOH and ACN). This approach — the explanation of selectivity changes when modifier is changed in the mobile phase — has an additional important advantage. The application of this approach leads to an easier and simpler prediction of selectivity changes because molecular interactions of the solute with the components of the stationary phase only are considered. Although these interactions in the mobile phase are omitted, this approach allows the prediction of relative retention changes of solutes with no ambiguous interpretation of these changes, which is the case when interactions in both phases are considered. Additionally, the modifier selectivity changes on the molecular level are comprehensible, which can lead to more effective optimization of resolution.

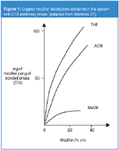
Figure 1
In the next section we will describe examples that confirm the approach postulated, based on data previously published and recent experiments.
Experimental
Solutes (Table 1) were obtained from different sources. All solvents and chemicals were of analytical grade. Water was bidistilled. Measurements of retention were performed with a HP 1050 liquid chromatograph (Agilent Technologies, Wilmington, Delaware, USA) equipped with 20 μL sample injector (Rheodyne, Cotati, California, USA) and a variable UV detector (HP-1050) operating at 254 nm.


Table 1: List of solutes investigated.
Barbiturates and phenols were analysed using a stainless-steel column (4.6 mm × 100 mm), which was packed with Lichrosorb RP-18, Si 100 (Merck KGaA, Darmstadt, Germany) after silanization with HMDS.22 Phenolic acids were analysed using a Zorbax SB C-18 (4.6 mm × 150 mm, 5 μm, Agilent Technologies) column. Hold-up volume was determined by the injection of pure water. The mobile phase comprised acetic acid buffer (0.02 M) at pH 4.6 when the retention of phenolic acids was determined. In the case of all remaining solutes the eluents contained 0.1% acetic acid for suppressing dissociation of acidic compounds and residual silanol groups.
Results and Discussion
The following discussion of the relative retention changes of solutes investigated is based on their abilities to form molecular interactions with modifier molecules in the stationary phase region of RP-HPLC systems. Properties of the modifier molecules for these interactions are expressed in a solvent selectivity triangle, which was proposed by Snyder, and modified using normalized solvatochromic Kamlet–Taft parameters (Figure 2 ).13,14

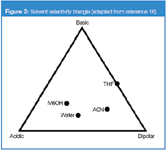
Figure 2
In this triangle modifiers are grouped according to their ability to donate or accept protons in hydrogen bond interactions and to form dipolar interactions. MeOH is characterized by its high propensity to interact as a proton donor (hydrogen bond acidity is equal to 0.93), a strong proton acceptor (hydrogen bond basicity = 0.62) and a relative high ability to form dipolar interactions (dipolarity/polarizability = 0.60). ACN possess the largest dipole moment of the three modifiers considered. Its Kamlet–Taft dipolarity/polarizability parameter is 0.75.
However, ACN is characterized by a weak ability to be a proton acceptor (hydrogen bond basicity = 0.31) and much weaker as a proton donor (hydrogen bond acidity = 0.19). THF (hydrogen bond basicity = 0.55) accepts protons more strongly than ACN, but its ability to form dipolar interactions (dipolarity/polarizability parameter = 0.58) is lower than ACN and it shows no proton donor properties (hydrogen bond acidity = 0.00).
Modifier Change
ACN versus THF: It can be expected that solutes possessing proton donor groups should increase their retention relative to solutes with electron donor groups in the THF system compared with ACN. This assumption corresponds with experimental results. As can be seen from Figure 3 the correlation line for phenols (solutes with a proton donor group) lies above that line for substances with electron donor groups.19
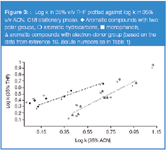
Figure 3
A similar dependence is shown in Figure 4 for barbiturates, phenols and benzene derivatives with an electron donor group. In this instance the correlation line for phenols is above the line for solutes with a proton acceptor (electron donor) group, whilst the line for barbiturates lies above the line for phenols. The barbituric acid derivatives are ureids of malonic acid. Their molecules (if not substituted at nitrogen atom) can possess either imid groups or phenol groups as a result of enolysis. So barbiturate molecules can show strong proton-donor properties in H-bond formation. The interaction of two imid groups in a barbiturate molecule is stronger than for monophenols. Hence, their correlation line is located above the line for phenols. It is worth noticing that points for barbiturates with a single imid group (hexobarbital, methylphenobarbital) are below the line for phenols.

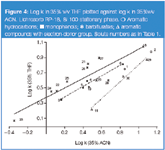
Figure 4
The greater ability of THF relative to ACN to act as a proton acceptor causes increased retention for phenolic acids with free and unshielded –OH groups (caffeic acid, 4-hydroxybenzoic acid, protocatechuic acid, 3-hydroxybenzoic acid, 4-coumaric acid) relative to substances which possess shielded –OH groups (sinapinic acid, veratric acid, wanilic acid ) in THF compared with an ACN system (Figure 5).

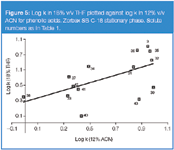
Figure 5
As we mentioned previously, the presence of one proton donor group in a solute molecule increases its retention relative to a solute without this group in a system with THF compared with ACN. Hence, the presence of two proton donor groups in the solute molecule should lead to an increase in specific interactions in the stationary phase, which makes the discussed effect stronger. This can be observed in Figure 3, where the retention of dihydroxynaphthalenes is increased relative to monophenols in the THF system compared with ACN. A similar effect, though less marked, is demonstrated for phenolic acids (Figure 5).
Increased retention of phenolic acids with two hydroxylic groups (protocatechuic acid, caffeic acid) relative to solutes with one –OH group (3-hydroxybenzoic acid, 4-hydroxybenzoic acid, 3-coumaric acid, 4-coumaric acid) is observed in the THF system compared with ACN. Similarly, the presence of two imid groups in a barbiturate molecule is responsible for its retention increase compared with a solute molecule with a single group in the THF system relative to ACN. (Figure 4).

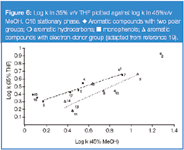
Figure 6
THF versus MeOH: MeOH shows a slightly greater ability to act as a proton acceptor than THF. However, sorption of MeOH in/on a C18 stationary phase is much lower21 than for THF. This effect implies that the stationary phase region in a system with THF participates more strongly in a retention rise for solutes with a proton donor group relative to those with a proton acceptor group, compared with the MeOH system. Indeed, in Figures 6–7 the retention increase can be observed for compounds with a proton donor group (phenols, barbiturates), compared with aromatic derivatives containing other polar groups, which have proton acceptor properties, in a THF system relative to MeOH.
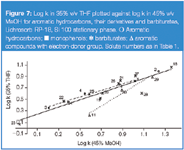
Figure 7
The effect is similar to that demonstrated for the THF system versus the ACN system discussed previously. According to our approach the increased retention of phenolic acids with unshielded proton donor groups compared with those containing shielded groups in the THF system is justified relative to the MeOH system (Figure 8). Methanol, in contrast to ACN and THF, is capable of interacting as a proton acceptor and a proton donor.
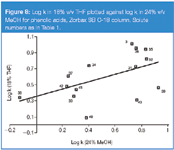
Figure 8
This property of MeOH allows molecular interactions with solute molecules possessing both proton-donor and proton-acceptor groups (e.g., barbiturates). These properties of the MeOH molecule can be applied to explain the location of the correlation line for barbituric acid derivatives relative to the phenol line, for the relationship THF versus ACN (Figure 4), compared with THF versus MeOH (Figure 7). The correlation line for barbiturates is below the phenols line (Figure 7), which is in contrast to the results in Figure 4. This behaviour demonstrates stronger molecular interactions of barbiturates relative to phenols with MeOH than with ACN in the stationary phase.

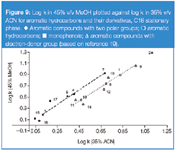
Figure 9
MeOH versus ACN: In Figures 9–10 the correlation of the retention of the investigated solutes in systems with MeOH and ACN is demonstrated. The correlation line for solutes with proton donor groups lies above the line for solutes with proton acceptor groups. The explanation of this effect stems from the greater ability of MeOH to be a proton acceptor than ACN. However, this effect is not as clearly demonstrated for THF versus MeOH or THF versus ACN systems. The explanation for this is related to the lower sorptions of ACN and MeOH in/on the stationary phase compared with THF. This effect is additionally demonstrated in Figure 10 by the line positions for barbituric acid derivatives relative to phenols.
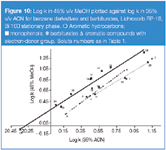
Figure 10
Ordering of the Stationary Phase
The previous discussions will now be complemented by considering the ordering of the hydrocarbon chains of the non-polar stationary phase caused by the organic component of the eluent. The degree of this ordering depends on the modifier type as mentioned previously. It means that solute molecules with a branched and/or bulky structure should demonstrate restricted entropic penetration of the more ordered stationary phase (such as the THF system compared with the less ordered stationary phases in the ACN or MeOH systems).
In Figures 3 and 6 good correlations between the retention factors for benzene derivates with one polar electron donor group for THF versus ACN and THF versus MeOH systems are shown, respectively. However, a few marked deviations can be observed. The points for nitrobenzene and benzonitrile are positioned above the correlation line. In contrast, the points for esters lie below this line. Nitrobenzene and benzonitrile have planar structure, while molecules of benzene derivatives with ester group(s) form a more bulky (branched) structure.19
A C18 stationary phase in the system with THF is more ordered than with ACN and, especially, with MeOH. Molecules with a branched (or bulky) structure should show lower entropic penetration of this stationary phase region relative to molecules of planar structure in the THF system compared with an ACN or MeOH system. This effect is analogous to the slit model of selectivity variations reported by Sander and Wise.23 According to this model, aromatic hydrocarbons of planar structure demonstrate relative retention increase compared with non-planar solutes in the RP system with the stationary phase with a higher coverage density relative to the stationary phase of lower coverage density. A similar effect is demonstrated in this paper for phenolic acids. The separation factors (α) in both MeOH and ACN systems for solute pairs: 4-coumaric acid and veratric acid; caffeic acid and syringic acid; 3-coumaric acid and sinapinic acid are in the range 1.00–1.14.
The separation factors of the solute pairs mentioned previously in the THF system are equal to 3.70, 3.43 and 2.70, respectively. This variation can be related to differences in the shapes of the molecules considered. Veratric acid, syringic acid and sinapinic acid have more branched molecular structures relative to their pair members mentioned 4-coumaric acid, caffeic acid and 3-coumaric acid, respectively.
Analogously, in Figures 4 and 7 the point location for 5-(4-methylphenyl)-5-phenylhydantoin relative to THF versus ACN and THF versus MeOH correlation lines for barbiturates is understandable. This solute is similar to barbiturates and possesses proton donor and proton acceptor properties, and additionally possesses two phenyl groups in position 5. This latter property is responsible for both the branched structure of the molecule and for a shielding effect against one of the two imid groups.
Hydrophobic Character of the Stationary Phase
Another feature of the investigated system is the change of hydrophobic character of the C18 stationary phase region when one modifier is replaced by another in the mobile phase. This effect may be discussed by comparing the retention data of benzene and toluene, which do not possess any polar groups. So these molecules can indicate a change of hydrophobic character of the stationary phase when their retentions are compared with other solute groups for the modifier systems investigated.
The location of points for these solutes, above the correlation line for phenols and barbiturates in Figures 6 and 7, indicates that benzene and toluene show increased retention relative to phenols and barbiturates in the THF system compared with the MeOH system. It means that the hydrophobic character of the stationary phase region with THF in the mobile phase is stronger than for MeOH. A comparison of the point location of benzene and toluene relative to correlation plots presented in Figure 9 leads to the conclusion that the stationary phase region in the system with ACN is more hydrophobic than for MeOH.
These points practically lay on the correlation plot for aromatic compounds with electron donor groups. In contrast, hydrophobicity of the stationary phase region in systems with THF and ACN seems to be similar. This remark is confirmed by the location of benzene and toluene points between the correlation line for aromatic compounds with proton donor groups and the correlation line for aromatics with electron donor groups (Figure 3).
Significantly greater sorption of THF and ACN compared with MeOH is responsible for higher hydrophobicity of the stationary phase region of the chromatographic systems with the first two modifiers. It should be noted that the hydrophobicity changes of the stationary phase region, when modifier is replaced in the eluent, could be especially useful for the separation of aromatic hydrocarbons from aromatic hydrocarbons with polar groups.
Change of Separation Factor
The influence of organic modifier on the separation selectivity discussed is also witnessed by variations in the separation factor, α2,1 (α2,1 = k2/k1) and expressed as log α2,1 values, presented in Table 2 (subscripts are attributed to solutes in the table). The presence of the –OH group in the solute molecule leads to a retention increase of this solute relative to the solute with the electron-donor group in the THF system compared with ACN or MeOH systems.

Table 2: Values of separation factors of some pairs of solutes.
The following pairs of solutes can be listed as evidence of this effect: 4 methylphenol (2) and acetophenone (1) (log α 2,1is equal to 0.33 in the THF system, while in MeOH and ACN log α 2,1is equal to –0.02 and –0.07 respectively), 2-ethylphenol (2) and anisole (1) (log α 2,1 is equal to 0.24 in the THF system, while in MeOH and ACN it is 0.01 and –0.04 respectively) and 4-methylphenol (2) and methylbenzyl ketone (1) (log α 2,1is equal to 0.33 in THF system, while in MeOH and ACN it is 0.02 and –0.15 respectively.
The presence of two –OH groups in the solute molecule leads to retention changes relative to a solute with one –OH group. 1,5-dihydroxynaphthalene is characterized by stronger retention relative to monophenols (e.g., phenol and 2-naphtol) in the THF system compared with ACN or MeOH. The value of log α 2,1for the solute pair 1,5-dihydroxynaphthalene (2) and phenol (1) is equal to 0.18 in the THF system, -0.10 in the MeOH system and 0.00 in the ACN system. For the pair of solutes 2-naphtol (2) and 1,5-dihydroxynaphthalene (1) the log α 2,1value in the THF system is 0.30, while in the MeOH system it is 0.84 and 0.67 in the ACN system.
The retention decrease of 1,2-dinitrobenzene (2) relative to 1,4-dinitrobenzene (1) in the THF system is expressed by log α 2,1 value of -0.13, while the respective values of log α 2,1 values are 0.04 in the ACN system and 0.03 in the MeOH system. This behaviour can be explained with regard to the difference in planarity of these molecules. The planar structure of 1,2-dinitrobenzene is disturbed by the vicinity effect of two nitro groups. The ordered stationary phase with THF demonstrates lower entropic accessibility towards molecules of this compound compared with 1,4-dinitrobenzene.
The presence of electrostatic interactions between nitrobenzene and THF and planarity of the former molecule explain the increase of nitrobenzene (1) retention compared with methyl phenyl acetate (2)19 in THF system relative to ACN or MeOH systems (log α 2,1values for THF = -0.15; ACN = 0.02, MeOH = 0.10). The retention increase of nitrobenzene relative to methyl phenyl acetate in the ACN system compared with MeOH can be explained by the greater ability of ACN to form dipolar interactions in the stationary phase.
The log α 2,1values of solute pair dimethyl isophthalate (2) and methylbenzoate (1) decrease in following order of the modifier systems: MeOH (0.13), ACN (0.06), THF (0.02) systems. Increase of ordering of a surface region of the staionary phase is also represented by the same sequence of these modifier systems. This means it should be clear that relative retention of the solute with the more highly branched molecule structure diminishes relative to the one with a less branched structure according to the order of the systems presented.
Reversed-Phase and Normal-Phase HPLC Systems
The modifier selectivity changes in RP-HPLC systems are similar to selectivity variations in normal-phase (NP) systems. It was reported that the retention increase of benzene derivatives with proton-donor groups relative to benzene derivatives with electron-donor groups was observed in the system with a silica–binary eluent composed of dioxane and heptane compared with the system composed of silica–THF plus heptane (or ethyl acetate plus heptane).24 A dioxane molecule, with two ether oxygen atoms, can form a hydrogen bond between one oxygen atom and a silanol group from the silica surface while, the second oxygen atom can be directed to the mobile phase and interacts with a solute molecule possessing a proton-donor group.
In contrast, a THF molecule has one oxygen atom and cannot form a hydrogen bond with a solute molecule possessing a proton-donor group when the former is adsorbed on the silica surface, as the dioxane molecule does. In Figure 11 the chromatograms of an artificial mixture of solutes with proton donor and proton acceptor groups are presented. In this figure the discussed effect can be easily distinguished (phenols show increased retention relative to remaining solutes in the dioxane system compared with the THF one). Another example of this relationship has also been presented for planar chromatography systems.25

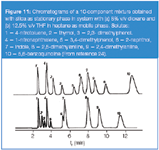
Figure 11
Conclusions
The organic modifier type in the mobile phase of a RP-HPLC system can lead to considerable selectivity changes. These selectivity changes can be predicted by understanding both the molecular interactions of the solute molecules with modifier molecules in the region of the RP-HPLC stationary phase surface and by the ordering of the stationary phase with respect to the modifier type used in the mobile phase.
This explanation for selectivity changes negates the molecular interactions occurring in the mobile phase. This means that selectivity change prediction is simplified and removes confusion arising when the relative retention changes are interpreted in terms of the molecular interactions of the solutes in both mobile and stationary phases.
This approach can be useful for optimizing separations of sample mixtures and for a better understanding of molecular mechanisms in RP-HPLC separations.
Anna Klimek-Turek is PhD student at Medical University of Lublin, Poland, her research interests include retention mechanism and solvent selectivity in reversed-phase and normal-phase liquid chromatography (HPLC) and optimization of separation of biologically active compounds by liquid chromatography (LC).
Tadeusz H. Dzido is head of Department of Physical Chemistry at Medical University of Lublin, Poland, his research interests include retention mechanism and solvent selectivity in reversed-phase and normal-phase liquid chromatography (HPLC and TLC), optimization of separation of biologically active compounds (LC), planar electrochromatography (PEC and PPEC) — optimization of separation and construction of the devices for PEC.
Heinz Engelhardt is professor emeritus at University of Saarland, Germany, his research interests include liquid chromatography, capillary electrophoresis and electrochromatography, supercritical fluid chromatography, stationary phases for liquid chromatography methods and separation of different compound types.
References
1. R. Kaliszan (Ed.), Structure and Retention In Chromatography. A Chemometric Approach, (Harwood Academic Publishers, Amsterdam, 1997).
2. K.B. Sentel and J. G. Dorsey, Anal. Chem., 61, 930 (1989).
3. A. Zdrouen et al., LCGC International, 5, 28 (1992).
4. T.H. Dzido and E. Soczewinski, J. Chromatogr., 550, 71–76 (1991).
5. T.H. Dzido and H. Smolarz, J. Chromatogr. A, 679, 59–66 (1994).
6. T.H. Dzido and A. Sory, Chem. Anal., (Warsaw), 41, 113 (1996).
7. V. Harang et al., Chromatographia, 54(11–12), 703–709 (1994).
8. W. Melander and C.S. Horvarth, in High Performance Liquid Chromatography Advances and Perspectives Volume 2, C.S. Horvarth, Ed, (Academic Press, New York, USA, 1980).
9. C.S. Horvath and W.Melander, J.Chromatogr. Sci., 15, 393–400 (1977).
10. R.P.W. Scott, J. Chromatogr. A, 656, 51 (1979).
11. F. Murakami, J. Chromatogr, 178, 393 (1979).
12. J. Ko and J. C. Ford, J. Chromatogr. A, 913, 3 (2001).
13. L.R. Snyder, P.W. Carr and S. C. Rutan, J. Chromatogr. A, 656, 537 (1993).
14. W. Kiridena, F.C.Poole, J.Planar Chromatogr., 12,13–25 (1995).
15. U.D. Neue, A. Mendez, J. Sep.Sci., 30, 949–963 (1993)
16. T.H. Dzido, J.Liq.Chrom. & Rel.Technol., 23(18), 2773–2788 (2000).
17. E.H. Slaats, Z.S. Heemstra and H. Poppe, Study of The Mobile Phase Interactions and Their Influence on the Retention and Selectivity in RPHPLC by Means of Solute Activity Coefficient, Measured in Methanol-Water, Acetonitrile-Water and Tetrahydrofuran-Water Solvent System" in Influence of mobile Phase and Stationary Phase Compositions on the retention in High Performance liquid chromatography (University of Amsterdam, Amsterdam, 1980) pp. 21–49.
18. D. Bolliet and C. F. Poole, Analyst, 123, 295–299 (1998).
19. T.H. Dzido, T. E. Kossowski and D. Matosiuk, J. Chromatogr. A, 947,167. (2002).
20. B.K. Lavine, J. P. Ritter, S. Peterson J. Chromatogr. A, 946 83–90 (2002).
21. R.M. McCormick and B. L. Karger, Anal. Chem., 52, 2249 (1980).
22. T.H. Dzido and H. Engelhardt, Chromatographia, 39,(1/2) July (1994).
23. L.C. Sander, S. A. Wise, J. C 656, 537 (1993).
24. T.H. Dzido, E. Soczewifski, J. Chromatogr., 388, 99–104 (1987).
25. T.H. Dzido and B. Polak, J. Planar Chromatogr., 6, 378–381 (1993).















New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.




