Determination of Acrylamide in Water by Liquid Chromatography Coupled to Tandem Mass Spectrometry
An analytical method based on liquid chromatography coupled to tandem mass spectrometry (LC–MS–MS) has been developed for the determination of acrylamide in water. To obtain clean extracts and low detection limits, an activated carbon cartridge was investigated for use in solid-phase extraction (SPE), and extraction conditions such as desorption solvent and elution volume were optimized by a series of experiments. High recoveries (99.1–99.8%) were obtained using the activated carbon solid-phase extraction cartridges with methanol as the eluent. This method could be applied to the quantification of acrylamide in environmental water samples.
An analytical method based on liquid chromatography coupled to tandem mass spectrometry (LC–MS–MS) has been developed for the determination of acrylamide in water. To obtain clean extracts and low detection limits, an activated carbon cartridge was investigated for use in solid-phase extraction (SPE), and extraction conditions such as desorption solvent and elution volume were optimized by a series of experiments. High recoveries (99.1–99.8%) were obtained using the activated carbon solid-phase extraction cartridges with methanol as the eluent. This method could be applied to the quantification of acrylamide in environmental water samples.
Acrylamide (1), a known neurotoxin and putative human carcinogen, has been included among the substances to be monitored in drinking water. This compound has been regulated by the European Council Directive 98/83/EC (2) with a minimum quality requirement of 0.1 µg/L in drinking water. The main source of acrylamide to drinking water is the release of residual monomer from polyacrylamide coagulants used as a clarifier in raw water treatment.



As a result of its high solubility in water (2155 g/L at 30°C) and its low levels in water, acrylamide is not easy to detect. There are very few reports on the determination of acrylamide in potable water At present, a frequently used method for the analysis of acrylamide relies on analyte derivatization and gas chromatographic (GC) separation (3). However, derivatization is often considered as time consuming, laborious, and can lead to a potential loss of analyte because of unstable or incomplete derivatization. Several groups have described a range of methods to quantify acrylamide by direct injection and reversedphase ultraviolet high performance liquid chromatography (HPLC–UV) with a limit of detection (LOD) of 5 µg/L (4) and by solidphase extraction (SPE) and gas chromatography coupled to mass spectrometry (GC–MS) analysis (5) to give limits of detection here. However, these methods are not sensitive enough for the analysis of low levels of acrylamide in water. The development of sensitive and reliable analytical methods for the quantification of acrylamide in potable water was considered as essential. The current study found that MS coupled to LC (6), using labelled acrylamide as the internal standard (IS), was now the most appropriate technique for the determination of acrylamide in water.
The purpose of this study was to develop a method for the determination of acrylamide in water at low levels. A reversed-phase LC–MS method based on a stable isotope dilution assay was developed for acrylamide analysis. Effectiveness of the enrichment process using the activated carbon cartridge for SPE was evaluated, and parameters such as desorption solvent and elution volume were optimized.
Experimental
Materials and Chemicals: Acrylamide (>99%) was provided by Dr. Ehrenstorfer GmbH, 13C3-acrylamide was purchased from Cambridge Isotope Laboratories. All organic solvents were of HPLC-grade quality. Stock solutions of acrylamide and 13C3-acrylamide were prepared by dissolving the compounds in methanol. These solutions were then appropriately diluted with water to prepare working standards.
The carbon SPE cartridges were from Varian (500 mg, 6 mL); Oasis MCX (150 mg, 6 mL) were from Waters; Supelclean ENVI-18 (500 mg, 6 mL) and Carb/NH2 (500 mg, 6 mL) were from Supelco. Activated carbon was from Sinopharm Chemical Reagent Co.,Ltd.
Nitrocellulose syringe filters of 0.45 µm were purchased from Xiboshi. The water used was purified (18 MΩ quality) by a Milli-Q system (Merck Millipore).
River water was collected from Xiangjiang River.
Chromatographic Conditions: The chromatographic separation of acrylamide was performed by reversephase LC using a 2.1 mm × 150 mm, 3.5-µm XBridge column (Waters). Optimum separation was achieved with an isocratic elution using methanol:water at a ratio of 10:90 as mobile phase at a flow rate of 0.2 mL/min. The sample volume injected was 10 µL.
MS Detection: MS–MS detection was performed on a TSQ Quantum Ultra triple quadrupole instrument (Thermo Fisher Scientific). Electrospray ionization (EI) in the positive ionization (PI) mode was used. The optimal source working parameters for monitoring positive ions were: Spray voltage: 4000 V; capillary temperature: 350 °C; Collision energy: 10 eV; sheath gas: 35 a.u; auxiliary gas: 10 a.u.
The MS detector operated in selected reaction monitoring (SRM) mode at m/z 72 and 75 for acrylamide and labelled 13C3-acrylamide, and fragment ion at m/z 55 and m/z 58 were used for peak identity confirmation.
Sample Preparation: Prior to the extraction stages, samples were filtered through 0.45-µm nitrocellulose syringe filters. The cartridges were conditioned with 5 mL methanol followed by 5 mL of water. Each cartridge was loaded with 100 mL of filtered water, and then dried by nitrogen for 5 min. The target compounds collected on the cartridges were eluted with 10 mL of methanol. The extract was evaporated under a stream of nitrogen. Finally, acrylamide was dissolved in 1 mL of water and filtered as before and transferred into amber glass vials for LC–MS–MS analysis.
Quantification: Acrylamide was quantified using a linear calibration function that was established with standard solutions of acrylamide at concentration levels 1.00 µg/L, 5.00 µg/L, 10.0 µg/L, 50.0 µg/L, and 100 µg/L. In all instances, 5.0 µg/L of 13C3-acrylamide internal standard was used for isotopic dilution quantification. The addition of a known level of internal standard to the samples at the beginning of the extraction process allowed the quantification of the analyte. This method gave more precise results because a correction of both extraction efficiency and changes in instrument performance were achieved.
Results and Discussion
Mass Spectrum of Acrylamide: An HPLC–MS–MS analytical method has been developed for the determination of acrylamide in water. The system was operated in positive electrospray and selected reaction monitoring mode. The detected ions of analyte and internal standard were [C3H5NO]+ = 72 and [13C3H5NO]+ = 75, respectively. The daughter ions at 55 m/z and 58 m/z were acquired by the loss of a protonated amine in acrylamide and 13C3-acrylamide, respectively. The dwell time was 1 s. An increase in dwell time led to a loss in linearity without a significant increase in sensitivity (7). The chromatograms of acrylamide and 13C3-acrylamide are shown in Figure 1.

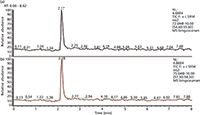
Figure 1: Chromatograms of (a) acrylamide and (b) 13C3-acrylamide with a concentration of 5.0 µg/L.
Optimization of SPE Procedure:
The Option of the Cartridge: A recovery assay was performed by adding 0.02 µg/L, 0.05 µg/L, and 0.10 µg/L of acrylamide to assess the extraction value. Five different SPE-cartridges with 10 mL methanol elution solvents in series were studied.
The results are shown in Figure 2. Activated carbon had the highest recovery for this application. The activated carbon has been frequently used in SPE of the polar micromolecules because of its wide specific surface area as well as reliable collection efficiency. Kawata et al. (8) reported the use of a granular activated carbon fibre as a device for SPE of eight hydrophilic organic compounds in environmental water. From these results, the activated carbon cartridge was selected as the extraction cartridge.

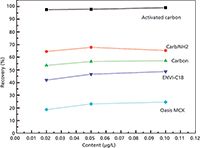
Figure 2: The recoveries of acrylamide with different cartridges.
Desorption Solvent: Desorption efficiencies for the target compound were determined by passing 100 mL of purified water spiked with 0.005 µg of the compound and 13C3-acrylamide through the activated carbon cartridges. After the cartridges were dried, the compounds were eluted by using methanol, 95% methanol, 90% methanol, acetonitrile, and chloroform. The results are presented in Table 1.


Table 1: A comparison of different solvents on acrylamide recovery in water.
The recoveries of 95% elution solvent reached 99.7% and were the highest. The performance of chloroform was the worst; it had only 49.8% recovery. The results matched the polar order of the solvents well. Because of the high polarity of acrylamide, water is a good choice as an elution solvent. However, as shown in Table 1, higher amounts of water did not significantly improve the recoveries, and instead prevented fast evaporation in the enrichment step. Therefore, methanol was recommended for the desorption solvent of the compounds from the extraction cartridge. Moreover, in SPE the elution volume is an important feature to take into account. The effects of different volumes of methanol on the recovery were studied. The elution curve (recovery [%] versus volume of elution) was established (Figure 3). As can be seen from the curve, the recoveries of acrylamide improved very little when the elution volume was more than 10 mL. For this reason, the optimum volume needed to remove the retained acrylamide quantitatively was set at 10 mL.

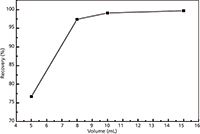
Figure 3: The effect of elution volume on the recovery of acrylamide.
Method Performance: To check the performance of the method quality, parameters such as LOD, repeatability, and linearity range were established. The results are shown in Table.2.

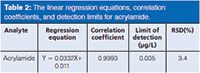
Table 2: The linear regression equations, correlation coefficients, and detection limits for acrylamide.
LOD was determined as the amount of analyte that produced a signal-to-noise ratio (S/N) of 3:1, and was calculated using a blank sample (100 mL) spiked with very low amounts of acrylamide at the beginning of the extraction. The LOD value was 0.005 µg/L for a 100 mL sample. Repeatability was given as the relative standard deviation (RSD). Excellent repeatability of less than 3.4% was obtained for acrylamide.
The recovery yields of acrylamide in water were investigated with 0.02 µg/L, 0.5 µg/L, and 1.0 µg/L of standards compound in the water (blank). The results are shown in Table 3.

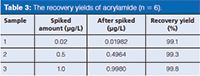
Table 3: The recovery yields of acrylamide (n = 6).
Sample Analysis: This method was applied for the determination of acrylamide in waters from Xiangjiang River. The results showed that acrylamide content of the water was lower than the LOD. Therefore, the concentrations of acrylamide in Xiangjiang River were estimated to be at an extremely low risk.
Conclusions
A method for the analysis of acrylamide has been developed. The method performs well at determining acrylamide in water where isotopically labelled internal standards are used. An improved purification and enrichment procedure was applied for the determination of acrylamide in water; recovery reached 99.8% and RSD was lower than 3.4%. This method could be applicable to the determination of acrylamide in water. Activated carbon is a very useful material and is widely used in environmental purification, such as air purification, sewage treatment, and solvent recovery,
Feng Jia-li is a senior technologist in food risk monitoring.
Chen Dong-yang is a technologist in food and water quality analysis.
Wang Yi-hong is an associate senior technologist in water quality analysis.
Li Bang-rui is a technologist-in-charge of poisoning factor research.
Zeng Dong is a technologist-in-charge of food risk monitoring.
Zhang Hao is a technologist-in-charge of harmful material detection in food.
Ding Li is an associate senior technologist in harmful material detection in food.
References
(1) International Agency of Research on Cancer Monographs (IARC), vol.60, 1994, 389.
(2) European Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption, European Union, Brussels, Off. J. Eur. Commun. L330 (1998) 32.
(3) E. Tareke, P. Rydberg, P. Karlsson, et al., J.Agric.Food Chem. 50(17), 4998–5006 (2002).
(4) M. Weideborg, T. Kallqvist, K.E. Odegrad, L.E. Sverdrup, and E.A. Vik, Norway. Water Res 35(11), 2645–2653 (2001).
(5) A.F. Lagalante and M.A. Felter, J.Agric.Food Chem. 52(12), 3744–3748 (2004).
(6) E. Bermudo, E. Moyano, L. Puignou, and M.T. Galceran, Talanta 76(2), 389–394 (2008).
(7) S. Cavalli, S. Polesello, and G.Saccani, J.Chromatogr.A 1039 (1–2), 155–159 (2004).
(8) K. Kawata, T. Ibaraki, A. Tanabe, et al., J. Chromatogr. A 911(1), 75–83 (2001).
(9) E. Bermudo, E.Moyano, L.Puignou, et al., Analytica Chimic Acta 559(2), 207–214(2006).







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")







