Data Integrity Focus, Part VII: A Data Life Cycle for Chromatography
LCGC North America
Several regulatory data integrity guidance documents require that laboratories understand the data life cycle of their processes, but provide only vague definitions of what a data life cycle is. Here is a practical interpretation.
Several regulatory data integrity guidance documents require that laboratories understand the data life cycle of their processes. Vague definitions of a data life cycle are presented in these documents. What is a practical interpretation?
This is the seventh in a series of articles addressing data integrity. The first article presented and discussed a Data Integrity Model to present the scope of data integrity and data governance program for an organization (1). The second looked at data process mapping to identify data integrity gaps involving a Chromatography Data System (CDS) ,and looked at ways to remediate them (2). The CDS was operated as a hybrid system which was the subject of the third article (3), and the fourth focused on how raw data and complete data mean the same thing for records created in a chromatography laboratory (4). The fifth part discussed how the updated USP <1058> general chapter on Analytical Instrument Qualification (AIQ) can help with ensuring data integrity with a Chromatography Data System (CDS) (5). In the sixth article, we examined the Quality Assurance (QA) oversight necessary to ensure data integrity (6). In this part, we look at the data life cycle that is mentioned in many of the data integrity guidance documents issued by regulatory bodies (7–9), and derive a relevant one for a chromatography laboratory.
Definition of a Data Life Cycle
One definition of a data life cycle is presented in the 2018 Medicines and Healthcare Products Regulatory Agency (MHRA) GXP data integrity guidance (8):
All phases in the life of the data from generation and recording through processing (including analysis, transformation or migration), use, data retention, archive/retrieval and destruction.
This is simple enough, and means managing all data generated in an analysis from life to death. There is also an explanation to the definition, stating:
Data governance ...... must be applied across the whole data lifecycle to provide assurance of data integrity. Data can be retained either in the original system, subject to suitable controls, or in an appropriate archive (8).
Data governance, in turn, is defined in the same document as:
The arrangements to ensure that data, irrespective of the format in which they are generated, are recorded, processed, retained, and used to ensure the record throughout the data lifecycle (8).
Scope of Data Governance
Now, we must consider what "the arrangements" mean in the context of governance of a data life cycle. In Part I of this series, a data integrity model was presented (1), and the layers of this model can be considered as "the arrangements" for data governance, as follows:
- Foundation: The right culture and ethos for data integrity
This consists of management leadership, development of an open culture, data integrity procedures and training, good documentation practices, and the ability to own up to mistakes, as well as assessment and remediation
- Level 1: Right instrument and system for the job
This includes integrated analytical instrument qualification and computerized system validation, so that instruments and systems are fit for their intended use.
- Level 2: Right analytical procedure for the right job
This refers to the development and validation of analytical procedures that are appropriate for the assay undertaken, and are validated under actual conditions of use.
- Level 3: Right analysis for the right job
This refers to the application of the lower layers in the analysis of samples to ensure that absolutely all data are acquired, interpreted, and calculated to final results in an open and transparent way. If a mistake is made, the analysis stops, as required by the FDA out of specification (OOS) results guidance (10). Included at this level are change control requests, deviations, and investigations, where appropriate.
All aspects of this data integrity model are components of data governance within a pharmaceutical quality system.
In this article, we will interpret the definition of data life cycle to develop a practical interpretation for chromatographers working in regulated laboratories. Please remember that a big problem with interpreting guidance documents and regulations is that one size rarely fits all situations. Therefore, we need to consider a flexible approach to an analytical data life cycle.
Data Process Mapping
In Part II of this series, we looked at data process mapping, and focused on just the analytical and calculation portions of a chromatographic process (2). The principles outlined in that article should be used as the basis for the discussions here, but extended to include sample management to the reportable result, as well as storing the data generated from a chromatographic analysis in electronic form linked with any paper records until the end of the record retention period.
Existing Data Life Cycles
There are two life cycles that have been derived from the regulatory guidance document definition above. The first is contained in the good automated manufacturing practice (GAMP) Guide on Records and Data Integrity, and consists of five phases (11):
- creation
- processing
- review, reporting and use
- retention and retrieval
- destruction
When compared with the MHRA data life cycle definition (8), there does not appear to be much more detail than the original definition. From an analytical and chromatographic perspective, the GAMP data life cycle compresses some of the most critical parts of the life cycle, so as to minimize their importance (such as peak processing and second person review). For example, integration of chromatograms is the subject of great scrutiny during inspections of regulated laboratories, and has resulted in the issue of several warning letters and 483 observations (12–14). The Parenteral Drug Association (PDA) Technical Report 80 (15) has a large section devoted to the integration and interpretation of chromatographic data, illustrating both acceptable and unacceptable practices; this information is buried in the GAMP data life cycle.
An alternative concept of data life cycle was explained in the second edition of my book, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (16). The major difference between this and the GAMP model is the concept of phases. This data life cycle has two phases, an active and an inactive phase. The active phase comprises the following activities:
- data acquisition
- data processing
- generation of reportable result
- information use
- short term retention.
The inactive phase comprises the following activities:
- long term archive
- data migration
- data destruction
The active phase is where most of the work occurs, from acquisition to data use, and includes short-term retention. However, this is the shortest part of the life cycle. The inactive phase is where the data and records are stored for the record retention period, and is the longest part, and, in the case of electronic records, potentially the most problematic. A critique of this life cycle is that it is generic, and is not focused on the chromatography laboratory.
Are These Data Life Cycles Adequate?
While these two data life cycles are useful in expanding the regulatory guidance definitions, they are difficult to adapt to a laboratory situation. Here are the reasons:
- Insufficient detail for chromatography, as there is no mention of sampling or sample preparation.
- Analytical procedures are not the same. Some procedures are simple as observation (such as color tests), and others involve complex instrumental analysis (for example, spectroscopy and chromatography).
Therefore, an analytical data life cycle is required that is flexible enough to cope with different analyses performed throughout pharmaceutical development and manufacturing, such as analytical development, bioanalytical, and quality control testing.
Controlling the Data Life Cycle
We'll start the analytical life cycle discussion from the regulations, in this case the Principle of EU GMP Chapter 4 on documentation. Here, it states that there are two primary types of GMP documents, instructions and records/reports:
Instructions include procedures and protocols that give directions for performing certain operations, or instructions for performing and recording certain discreet operations respectively.
Records: Provide evidence of various actions taken to demonstrate compliance with instructions... (17).
In both a regulated and a quality environment, chromatographers must have a procedure to perform a task-either a standard operating procedure, an analytical procedure, or a protocol. These instructions control a business process, and state what must be done, as well as the data and records to generate.
This control element is missing from both the data life cycles described above (11,16).
Active and Inactive Phases are Essential
Next, there is the concept of phases to consider, as mentioned in my original data life cycle model (16). Over the records retention period, there is typically a handover of records from a laboratory to a records management group or a Good Laboratory Practice (GLP) archivist (18,19) at or after the completion of work. This transfer marks the transition from an active to an inactive phase. Here, the records reside for the records retention period until a complaint or audit inspection retrieval request is made, or until formal destruction. Control via procedures is also a requirement for the inactive phase of a life cycle.
Second-Person Review
Hidden in the third activity of the GAMP data life cycle is review (11). As this is a critical part of any data life cycle, it needs to stand out rather than be obscured with other life cycle tasks. Second-person review is the key to ensuring that work has been carried out correctly, and that unofficial testing or falsification of data have not occurred (8,20,21).
A Flexible Analytical Data Life Cycle
Let me summarize where we are in the discussion now. An analytical data life cycle should have the following features:
- control of any life cycle by an instruction (procedures, plan, and/or a protocol)
- an active phase, where the work is carried out, and the reportable result generated
- review of the work before the release of the result
- an inactive phase, where records are managed and maintained by a separate group
What does Chromatographic Analysis Consist of?
Let us focus on the most interesting portion of a data life cycle: chromatographic analysis. Set up the instrument, inject samples, collect data, and integrate the peaks. Simple! But wait, where does the sample come from? We don't just inject a solid sample into a chromatograph, do we? We need a sample preparation step to make a solid sample liquid. So let us think this through and ask, What are the stages of a chromatographic analysis? Here's a list:
- sampling and sample management
- sample preparation
- instrument and chromatography data system (CDS) set up
- sample analysis, including system suitability tests (SSTs)
- integration of SSTs, standards, quality controls, and samples (in that order [23])
- calculation of the reportable result(s)
- report or certificate of analysis (COA) generation.
This seems a reasonable process flow. At each stage, you can identify the data and information that must be captured, integrated, calculated, and transformed. When expanded fully, this process should define the analytical data life cycle for a chromatographic analysis.
When an analysis is complete, and the tester or testers have checked their work to ensure that all data and records have been recorded and processed correctly, a second-person review should take place. The purpose of this review is twofold:
1. To confirm that the correct instructions and sample were used, the procedures were followed, data were generated and processed correctly, and, if errors are found, the record set is returned to the tester to correct.
2. To check that no falsification or poor data management practices have occurred.
This gives rise to an active analytical data life cycle, as shown in Figure 1, which is more applicable to an analytical laboratory than the two data life cycle models discussed earlier. Furthermore, you can see how the analytical portion equates to a typical chromatographic analysis.


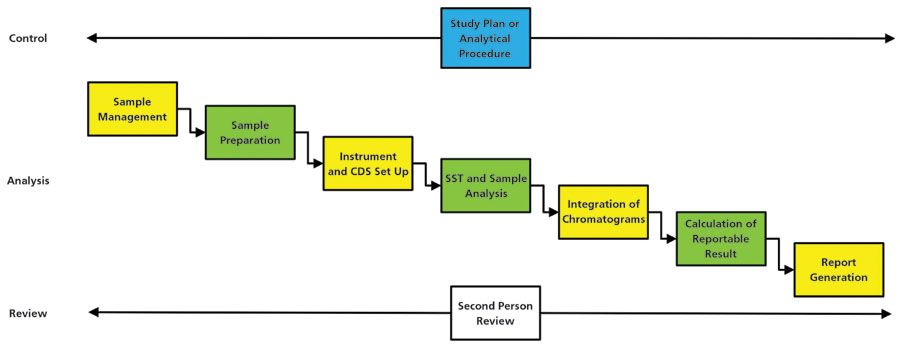
Figure 1: The active phase of an analytical data life cycle, adapted from reference (22).
One Size Fits All?
However, does a one-size analytical data life cycle fit all? Do all chromatographic methods need all stages? Let us consider the following scenarios:
- On-line or in process analysis may not require sample management or sample preparation.
- If the sample is liquid, sample preparation may not be required .
- Qualitative analysis will not require calculation of results.
Therefore, any analytical data life cycle model needs to be flexible to accommodate an individual analytical procedure, rather than a "one size fits all" approach. For more detail on this subject, there is further reading available (22,24).
Inactive Phase
Let us turn our attention to the least interesting (from a laboratory perspective), but most challenging part of a data life cycle: the inactive phase. As mentioned earlier, this is also the longest part of any data life cycle. First, we must consider the nature of the records that we could be managing here:
- Paper records, such as sample management and sample preparation, as well as instrument log book entries.
- Signed paper printouts linked with electronic records stored in the chromatography data system (hybrid system). This could also include printouts and spreadsheet files if the CDS is not used to calculate the reportable result.
- Electronically signed electronic records stored in the chromatography data system, or archived separately if the CDS has this function.
We will only consider the electronic records in the CDS either as hybrid or fully electronic, as paper alone is relatively easy to manage. As in the active phase of the analytical data life cycle, there must be instructions to control how the records are managed and handled throughout this phase. The inactive phase is shown in Figure 2.


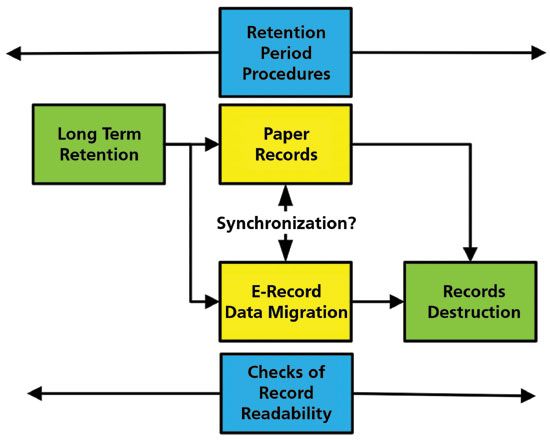
Figure 2: Activities in the inactive phase of an analytical data life cycle, adapted from reference (22).
Static and Dynamic Data
Chromatography data are dynamic; a trained user must interpret the chromatograms before calculation of the reportable result. In contrast, static data exists where the results are used as is, such as temperature results. The MHRA and FDA guidance documents both require dynamic data to be retained in dynamic format (8,21). This is a massive problem for the inactive phase of the life cycle with the current CDS systems. There is no easy way that all relevant data can be transferred from one supplier's system to another's, as there is no current universal data standard. Therefore, to achieve this regulatory expectation, either don't change your data system, create a CDS museum, or virtualize the old CDS to enable data to be reviewed. Printing to PDF is merely creating electronic paper and a static record.
Hybrid versus Electronic CDS Records
The simplest situation to consider is electronically signed electronic records (and therefore no printed paper). There should be a function within a CDS to lock the records when the reviewer signs, to state that the data are complete, consistent, and accurate. This means that the record set cannot be changed unless the electronic signatures are revoked or data are unlocked to perform further work (for example, a complaint is received after release of the product). The function to unlock the data set within the CDS must be restricted, and unlocking must be justified. If further work is performed, the advantage is that all updates of the data are held within a single environment, and audit trail entries monitor any change to the records.
In contrast, with a hybrid CDS, the electronic signature function will not be used, as the approval signature is applied outside of the system on the paper printout. Then the record set will not be locked by the system. This is a potential data vulnerability unless the data set can be isolated in or removed from the CDS. In the situation of a complaint as described above, the data set can be reviewed without the need to unlock the records, but if data are changed, there is now a second signed printout that must be managed. We now have electronic records in the CDS and two signed printouts to manage that must be synchronized, which is not the best way to manage data.
Data Migration Problems?
The record retention period for pharmaceutical products varies from batch expiry plus a year (25) to five years after release of the batch by the qualified person (17). Typically, companies will retain records for longer, due to product liability legislation. This means that the CDS holding electronic records for the record retention period will probably be updated at least once. Changes to the CDS application could affect, for example, integration algorithms, structure of the database (if used), and data file structure. It should be understood that moving from one version to another of the same CDS could involve data migration (16); the extent of that migration depends on the changes in the upgrade. Therefore, under the approved change request for the CDS, the impact on existing data in the system needs to be evaluated. For example, if the peak integration algorithm changes, what is the impact on previously acquired data?
If the inactive phase of the life cycle involves migration of electronic data, there is also a requirement to ensure that the migrated electronic records of hybrid systems are synchronized with the existing paper printouts.
Summary
Understanding the data life cycle and the data and records required to support complete data and raw data is essential for any data integrity program for a regulated laboratory. The data life cycle consists of an active and an inactive phase. Each phase must be controlled by instructions that determine what tasks must be done. The active phase involves sampling through to the generation of the reportable result by one or more testers. All records are then reviewed by a second person to bring the active phase of the life cycle to an end. The inactive phase of the life cycle lasts as long as the records retention period, and may include migration of electronic records in the CDS. If the CDS is a hybrid system, then the synchronization between the signed paper printouts and the underlying electronic records is essential.
References
(1) R.D. McDowall, LCGC North Am.37(1), 44–51 (2019).
(2) R.D.McDowall, LCGC North Am. 37(2), 118–123 (2019).
(3) R.D.McDowall, LCGC North Am. 37(3), 180–184 (2019).
(4) R.D.McDowall, LCGC North Am. 37(4), 265–268 (2019).
(5) R.D.McDowall, LCGC North Am. 37(5), 312–316 (2019).
(6) R.D.McDowall, LCGC North Am. 37(6), 392–398 (2019).
(7) MHRA GMP Data Integrity Definitions and Guidance for Industry (Medicines and Healthcare Products Regulatory Agency, London, United Kingdom, 2nd ed., 2015).
(8) MHRA GXP Data Integrity Guidance and Definitions(Medicines and Healthcare Products Regulatory Agency, London, United Kingdom, 2018).
(9) Good Practices for Data Management and Integrity in Regulated GMP /GDP Environments (Draft) (Pharmaceutical Inspection Co-operation Scheme, Geneva, Switzerland, 2018).
(10) FDA Guidance for Industry Out of Specification Results (Food and Drug Administration, Rockville, Maryland, 2006).
(11) GAMP Guide Records and Data integrity (International Society for Pharmaceutical Engineering, Tampa, Florida, 2017).
(12) FDA Warning Letter: Hubei Danjiangkou Danao Pharmaceutical Co. (Food and Drug Administration Silver Springs, Maryland, 2017).
(13) FDA Warning Letter: Divi's Laboratories Ltd. (Unit II) (Warning Letter 320-17-34), ( Food and Drug Administration Silver Springs, Maryland, 2017).
(14) FDA 483 Observations: Cadila Healthcare Limited (Food and Drug Administration, Rockville, Maryland, 2019).
(15) Technical Report 80: Data Integrity Management System for Pharmaceutical Laboratories (Parenteral Drug Association, Bethesda, Maryland, 2018).
(16) R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Royal Society of Chemistry Press, Cambridge, United Kingdom, 2nd ed., 2017).
(17) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation, E. Commission, Editor. 2011: Brussels.
(18) OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1, OECD Principles on Good Laboratory Practice (Organization for Economic Co-operation and Development, Paris, France, 1998).
(19) 21 CFR 58 Good Laboratory Practice for Non-Clinical Laboratory Studies (Food and Drug Administration: Washington, DC, 1978).
(20) WHO Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices (World Health Organization, Geneva, Switzerland, 2016).
(21) FDA Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers (Food and Drug Administration, Silver Springs, Maryland, 2018).
(22) R.D.McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry Press, Cambridge, United Kingdom, 2019).
(23) M.E. Newton and R.D. McDowall, LCGC North Am.36(5), 330–335 (2018).
(24) R.D. McDowall, Spectroscopy 33(9), 18–22 (2018).
(25) 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration: Sliver Spring, Maryland, 2008).
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")



. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

Determining the Effects of ‘Quantitative Marinating’ on Crayfish Meat with HS-GC-IMS
April 30th 2025A novel method called quantitative marinating (QM) was developed to reduce industrial waste during the processing of crayfish meat, with the taste, flavor, and aroma of crayfish meat processed by various techniques investigated. Headspace-gas chromatography-ion mobility spectrometry (HS-GC-IMS) was used to determine volatile compounds of meat examined.


