Data Integrity Focus, Part 1: Understanding the Scope of Data Integrity
LCGC North America
There are many factors to consider in a data integrity and governance program. Fortunately, a simple diagram can help us understand what needs to be covered.
Data integrity requires more than just ensuring that the calculated numbers of an analysis are complete, consistent and accurate. There is much more to consider. The full scope of a data integrity and data governance program can be presented and explained in a simple diagram.
Welcome to "Data Integrity Focus," a six part series on data integrity in regulated laboratories that will also be of use to other readers working under quality standards such as ISO 17025 (1). We will explore some selected topics in data integrity and data governance. To begin, we will discuss the scope of a data integrity program.
Last year in LCGC North America, Mark Newton and I wrote a six-part series on data integrity in the regulated chromatography laboratory (2–7) in which we reviewed the whole analytical process. In Part 1, we introduced briefly a four-layer model to explain the scope of data integrity (3). In this first part of "Data Integrity Focus," I would like to go into more detail of the model so that you can understand the different strands of a data integrity program. Note the use of the word "program." Data integrity has many strands of work; it is not a single project. There are multiple projects that come under the umbrella of a program. Let me explain the data integrity model in more detail so that you can see why.
Data Integrity Within a Quality System
Over the past decade, pharmaceutical regulation has focused on the development of a pharmaceutical quality system (PQS) based on the ISO 9000 quality management system (QMS) following the publication of the International Council for Harmonization (ICH) Q10 guidance (8) and the update of EU GMP Chapter 1 (9). In a PQS, senior management have overall responsibility and accountability for all activities and data generated (8,9). Although data integrity has always been implicit in regulations, section 1.9 of EU GMP Chapter 1 was updated so that work performed by Quality Control laboratories must proceed as follows:
“(iv) Records are made, manually and/or by recording instruments, which demonstrate that all the required sampling, inspecting and testing procedures were actually carried out. Any deviations are fully recorded and investigated (9).”
Implicit in this definition is that the records generated have adequate quality and integrity. As an aside, EU GMP Chapter 4 on documentation and Annex 11 for computerized systems are being revised to emphasize data integrity (10).
Data Integrity Model
To understand the scope of data integrity, a four-layer model has been developed, covering development, production, quality control (QC), and quality assurance (QA). The full GMP model is discussed in my books (11,12) and the initial discussion of the analytical portion was presented in Spectroscopy (13). The four layers are shown in Figure 1 and described below for a regulated laboratory and QA only:


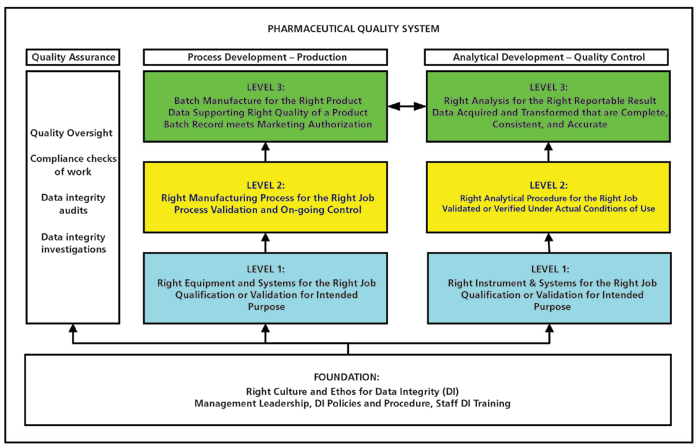
Figure 1: A Data Integrity Model. Reproduced with Permission from The Royal Society of Chemistry (11).
Foundation: Right Corporate Culture for Data Integrity
The foundation goes across all elements in an organization and is the data governance layer. The elements here for data integrity are management leadership, data integrity policies including data ownership, staff training in these procedures, management review including quality metrics and the establishment, and maintenance of an open culture with ethical working by all staff.
Level 1: Right Instrument or System for the Job
Analysis requires analytical instruments and computer applications to ensure data quality, and data integrity instruments must be qualified and software including spreadsheets must be validated. Included here are calibration, point-of-use checks, or system suitability test samples to confirm that the analytical instrument or laboratory computerized system is within user specifications before use.
Level 2: Right Analytical Procedure for the Job
For a laboratory, this is validation or verification of analytical procedures under actual conditions of use. What is not covered in current regulations or guidance is method development, which will determine the robustness of the procedure; this is the subject of a draft United States Pharmacopeia (USP) general chapter <1220> (14) on analytical procedure lifecycle management (APLM).
Level 3: Right Analysis for the Right Reportable Result
Here, process development and production provide the laboratory samples for analysis that are taken to demonstrate adequate product quality and conformance with the product specification in the marketing authorization (MA). It is this level where the work of the three layers below is essential for work to be performed ethically and correctly and where deviations occur they are investigated (9).
Quality Oversight
Although shown on the left of Figure 1 because of the sample link between production and quality control, the QA function is pervasive throughout the data integrity model to provide quality oversight of both production and laboratory operations, such as ensuring compliance with regulations, policies, and procedures as well as performing data integrity audits and data integrity investigations.
This is an overview of the data integrity model. We will now look in more detail at each level of the model for a regulated laboratory.
Foundation Level: Data Governance
The first level is called the Foundation Level for a very specific reason: Data integrity and data governance start with senior management involvement. Without it, any work at the levels above will be wasted. As shown in Figure 2, the Foundation Level has several elements that are essential for data integrity, which are explained below.

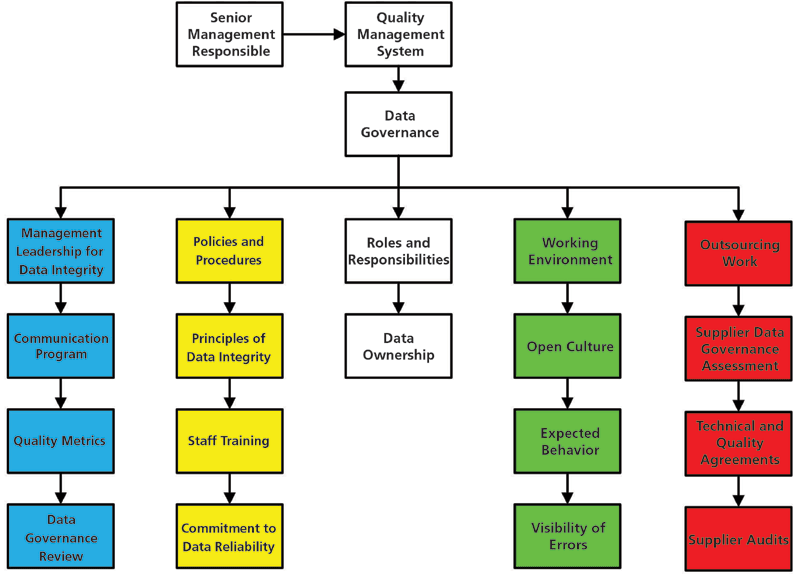
Figure 2: Data governance functions at the foundation level. Adapted from reference (11) with Permission.
Management Leadership and Involvement with the PQS
- Senior management leadership for data integrity that includes a communication program for all staff about the importance of data integrity and the impact it can have on patients and the organization if instances of noncompliance are found.
- Generation and use of quality metrics for monitoring data integrity. This is the subject of the two papers by Newton and McDowall (7,15) and will not be discussed further here.
- As part of the Pharmaceutical Quality System, a review of the effectiveness of the data governance and data integrity projects within the overall program. EU GMP Chapter 1 (9) mandates that management must review the QMS and data integrity is part of that review process.
Policies and Procedures Need to Be in Place, Including:
- Writing a data integrity policy with initial and ongoing training in its contents. After being trained, all members of staff should sign a commitment to data reliability for their work. Although the EMA Q&A (16) notes that there is no regulation for a data integrity policy, guidance documents issued by the Medicines and Healthcare Products Regulatory Agency (MHRA), the World Health Organization (WHO), and the Pharmaceutical Inspection Co-operation Scheme (PIC/S) guidances (17–21) all note that one is needed.
- Good documentation practices covering paper, hybrid, and electronic processes coupled with training in the procedure. This should include defining a flexible analytical data life cycle that was discussed in a recent column (22) with more detail of this approach in my book (11).
- Interpretation of analytical data, such as chromatographic integration, comparison of spectra using libraries, what are analysts allowed to do, and when and what specifically activities are prohibited.
- Training in these procedures with demonstrable understanding of the contents evidenced by using questionnaires or practical execution of the procedure.
Who Does What?
- The roles and responsibilities of all staff involved in a data governance and data integrity program including data ownership need to be documented and the information transmitted to all staff (11).
- Roles and responsibilities must be reinforced by appropriate sections in each individual's job or position description.
- Incorporation of data integrity goals into personnel objectives must be completed.
Quality Culture and the Working Environment
- Senior management needs to create an open quality culture where there are standards for expected behavior. This is probably the most difficult task in the whole data integrity program: It does not consist of a single e-mail, but an ongoing task to affect a culture change in an organization. It is not an event but a journey. ISPE has published a Cultural Excellence report (23) and there is also an abridged section on corporate culture in the recent GAMP Good Practice Guide on Data integrity – Key Concepts (24).
- Gemba walks where management gets to see issues in the laboratory first hand rather than filtered by subordinates (23). It is also an opportunity to influence staff directly by promoting the open culture and owning up to mistakes.
- Accordingly, there is an expected behavior and an open culture where mistakes can be admitted without blame. Admitting mistakes is also a regulatory expectation as noted in the Analyst Responsibilities section of the FDA OOS Guidance (25) that states:
"If errors are obvious, such as the spilling of a sample solution or the incomplete transfer of a sample composite, the analyst should immediately document what happened. Analysts should not knowingly continue an analysis they expect to invalidate at a later time for an assignable cause (i.e., analyses should not be completed for the sole purpose of seeing what results can be obtained when obvious errors are known)."
Outsourcing Work
- Any outsourced work requires an assessment of the outsourcing organization's or laboratory's data governance and data integrity status before technical and quality agreements are written and signed.
Level 1: Integrated Instrument Qualification and Computer Validation
There is little point in carrying out an analysis if an analytical instrument is not adequately qualified, or the software that controls it or processes data is not validated. Therefore, at Level 1, the analytical instruments and computerized systems used in the laboratory must be qualified for the specified operating range, and validated for their intended purpose, respectively. There are the following sources:
- USP <1058> for Analytical Instrument Qualification, 2018 version (26)
- GAMP Good Practice Guide for Validation of Laboratory Computerised Systems, 2nd Edition (27)
- Validation of Chromatography Data Systems, 2nd Edition (12)
These documents provide guidance and advice on these two interrelated subjects. Indeed, the new version of USP <1058> integrates instrument qualification and computer validation for analytical equipment (26) and the integrated approach is discussed in more detail in recent publications (12,28–30) A user requirements specification must be written for both instruments and software to define the intended use and against which the instrument will be qualified and the software validated. Where the software application must be configured to protect electronic records generated by the system, this must be reflected in the validation documents for the application software. By implementing suitable controls to transfer, mitigate, or eliminate any record vulnerabilities so that they can be adequately protected and ensure data integrity. Burgess and McDowall in an earlier LCGC series about an ideal chromatography data system (CDS) discussed some of the architecture, workflow and compliance requirements for ensuring data integrity (31–34).
Failure to ensure that an analytical instrument is adequately qualified or software is adequately validated means that all work in the top two levels of the data integrity model is wasted, as the quality and integrity of the reportable results is compromised by unqualified instrumentation and unvalidated and uncontrolled software.
Assessment, remediation, and long-term solution of paper processes and computerized systems are also included in this level of the model.
Level 2: Analytical Procedure Lifecycle Management
Using qualified analytical instruments with validated software, an analytical procedure is developed or established, and then validated or verified. The GMP requirement is that analytical methods must be verified under actual conditions of use as per 21 CFR 211.194(a)(2) (35), and, therefore, be fit for its intended use.
There are several published references for method validation from ICH Q2(R1) (36), FDA validation guidance documents (37,38) and the respective chapters in the European Pharmacopoeia (EP) and United States Pharmacopoeia (USP). However, the focus of these publications is validation of an analytical procedure that has been already developed. Method development is far more important, as it determines the overall robustness or ruggedness of any analytical procedure, but this process receives little or no attention in these publications. However, this analytical world is changing; following the publication in 2012 by Martin et al (39), there is a draft USP <1220> on The Analytical Procedure Lifecycle (14), issued for comment. This will mean a move from chapters focused only on validation, verification, or transfer of a method to a life cycle approach to analytical chapters that encompass development, validation, transfer, and continual improvement of analytical methods.
A life cycle approach to analytical procedures validation means that definition of an Analytical Target Profile (ATP) leads to good scientifically sound method development that ends with the definition of the procedure's design space, which now becomes important, as changes to a validated method within the validated design space would be deemed to be validated per se. There will be a transition period where the old approach is phased out while the new one is phased in. There is currently an ICH initiative that began in 2018 to update ICH Q2(R1) (36) to a life cycle approach (40).
Verification of Pharmacopoeial Methods
Given the vague descriptions of most analytical methods in various pharmacopoeias, it is amazing that any laboratory can get a method working at all. In essence, pharmacopoeial methods are unlikely to work as written. One of the reasons is that if a method for high performance liquid chromatography (HPLC) is developed using a specific supplier's C18 column, the only information about the column that appears in the monograph is a description of the packing and the column dimensions. For gradient methods, there is no information about whether the gradient is formed using a low-pressure or high-pressure mixing pump. For these reasons, analytical procedures based on pharmacopoeial "methods" need to be developed and verified under actual conditions of use as required by 21 CFR 211.194(a)(2) (35). The pharmacopoeia simply provides an indication of where to start but the details are left to the individual laboratory to develop, document, and verify.
Level 3: Analysis of Samples
Finally, at Level 3 of the data integrity model, the analysis of sample will be undertaken using the right analytical procedure, using a qualified analytical instrument and processing with validated software applications. To be successful, this also requires an open environment that enables data to be generated and interpreted, and the reportable result to be calculated, without bias or manipulation of data. Staff should be encouraged to admit any mistakes and there must be a no-blame culture in place based on the leadership of senior management from the foundation level of the model. It is also important not to forget the importance of the overall pharmaceutical quality system in providing the umbrella for quality such as the investigation of out-of- specification results, managing deviations and developing corrective and preventative actions. Figure 3 shows an analysis in practice and how the various levels of the data integrity model interact with each other. There are also the following elements of data governance:
- performing the work correctly and contemporaneously including any deviations
- effective and comprehensive second person reviews of the work
- visibility of errors
- investigation of aberrant results
- availability of both paper and electronic records for audit or inspection
- monitoring the development and maintenance of operational data integrity procedures and training.

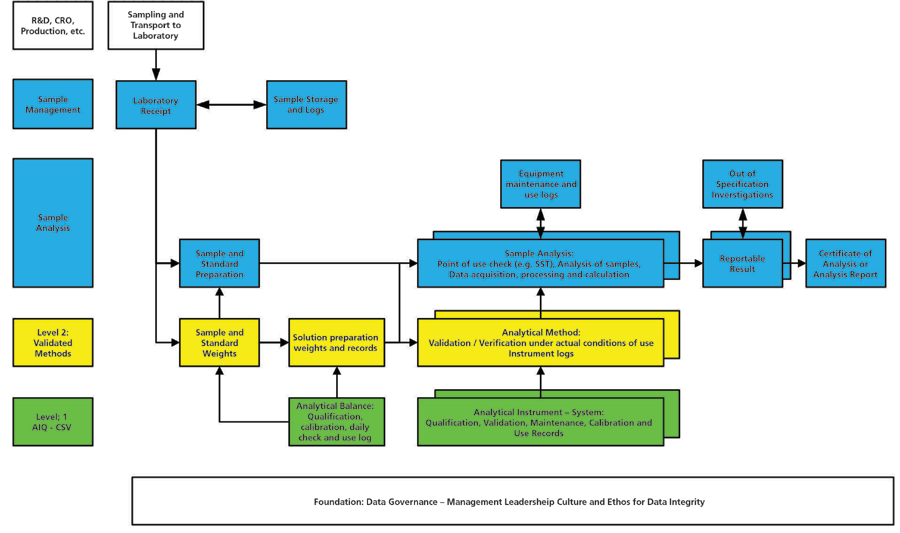
Figure 3: Interaction of the four levels of the data integrity model. Adapted with permission from reference (11). Definition of acronyms: Research and development (R&D), contract research organization (CRO), analytical instrument qualification (AIQ), computerized system validation (CSV), and system suitability test (SST).
These complete the laboratory levels of the data integrity model shown in Figure 3 but don't forget the quality oversight (checks of current work plus data integrity audits and investigations) shown in Figure 1.
Quality Does Not Own Quality Anymore
Figure 3 shows how the various levels of the laboratory data integrity model interact together. However, without the Foundation layer, how can the three other layers hope to succeed? The onus is on trained staff to act ethically. Also, without qualified analytical instruments and validated software, how can you be assured of the quality and integrity of the data used to calculate the reportable result? And so on up the levels of the model. It is less important where an individual activity is placed in the various layers; the primary aim of this model is to visualize for chromatographers and analytical scientists the complete scope of data integrity.
If the data integrity model works from the foundation through the three levels that exist on top, it means that the responsibilities for data integrity and data quality are now dispersed throughout the laboratory and organization, whilst the overall accountability for quality oversight remains with a quality assurance function. It is not the role of quality assurance to fix other people's mistakes. The responsibility for data integrity and data quality in the chromatography laboratory lies is with the analytical staff performing the work, showing that quality (that is, the quality control department) does not own quality anymore. Everyone in the laboratory and the whole organization does.
Data Integrity Guidances and the Data Integrity Model
When the material in the data integrity guidance documents from MHRA, FDA, EMA, WHO and PIC/S (Refs) are compared with the model, there are several gaps and there is no mention of:
- analytical instrument qualification and fitness for intended use in comparison with a heavy emphasis on control of computerized systems, nor
- analytical procedures, including robust method development and procedure validation.
All layers of the data integrity model are essential to ensure data integrity in a chromatography laboratory.
Summary
In this column, we have looked at a four-layer data integrity model to cover the whole scope of a data integrity program. The layers are interactive; ensuring data integrity depends on a foundation of data governance, qualified analytical instruments, and validated software with properly developed and validated robust analytical procedures. In the next article in this series, we will look at a way of identifying data integrity vulnerabilities in paper processes and computerized systems.
References
(1) ISO 17025-2017 General requirements for the competence of testing and calibration laboratories. 2017, International Standards Organization: Geneva.
(2) M.E. Newton and R.D. McDowall, LCGC North Am.36(5), 330–335 (2018).
(3) M.E. Newton and R.D. McDowall, LCGC North Am.36(1), 46–51 (2018).
(4) M.E. Newton and R.D. McDowall, LCGC North Am.36(4), 270–274 (2018).
(5) M.E. Newton and R.D. McDowall, LCGC North Am.36(7), 458–462 (2018).
(6) M.E. Newton and R.D.McDowall, LCGC North Am.36(8), 527–529 (2018).
(7) M.E. Newton and R.D. McDowall, LCGC North Am.36(9), 686–692 (2018).
(8) ICH Q10 Pharmaceutical Quality Systems. 2008, ICH, Geneva.
(9) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 1 Pharmaceutical Quality System. 2013, European Commission: Brussels.
(10) Work plan for the GMP/GDP Inspectors Working Group for 2018 2017, European Medicines Agency: London.
(11) R.D. McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories. (Royal Society of Chemistry Publishing, Cambridge, UK, 2019).
(12) R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Royal Society of Chemistry Publishing, Cambridge, UK, 2nd ed., 2017).
(13) R.D. McDowall, Spectroscopy,31(4), 15–25 (2016).
(14) G.P. Martin et al., Stumulus to the Revision Process: Proposed New USP General Chapter: The Analytical Procedure Lifecycle <1220> Pharmacopoeial Forum,43(1), 2017.
(15) M.E. Newton and R.D. McDowall, LCGC Europe, 30(12), 679–685 (2017).
(16) EMA Questions and Answers: Good Manufacturing Practice: Data Integrity. 2016; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/gmp_q_a.jsp&mid=WC0b01ac058006e06c#section9.
(17) MHRA GMP Data Integrity Definitions and Guidance for Industry 2nd Edition. 2015, Medicines and Healthcare products Regulatory Agency: London.
(18) MHRA GMP Data Integrity Definitions and Guidance for Industry 1st Edition. 2015, Medicines and Healthcare products Regulatory Agency: London.
(19) MHRA GXP Data Integrity Guidance and Definitions. 2018, Medicines and Healthcare products Regulatory Agency: London.
(20) WHO Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices. 2016, World Health Organization: Geneva.
(21) PIC/S PI-041 Draft Good Practices for Data Management and Integrity in Regulated GMP / GDP Environments. 2016, Pharmaceutical Inspection Convention / Pharmaceutical Inspection Co-Operation Scheme: Geneva.
(22) R.D. McDowall, Spectroscopy 33(9), 18–22 (2018).
(23) ISPE Cultural Excellence Report. 2017, International Society of Pharmaceutical Engineering: Tampa, FL.
(24) GAMP Good Practice Guide: Data Integrity - Key Concepts. 2018, International Society for Pharmaceutical Engineering: Tampa, FL.
(25) FDA Guidance for Industry Out of Specification Results. 2006, Food and Drug Administration: Rockville, MD.
(26) USP 41 General Chapter <1058> Analytical Instrument Qualification. 2018, United States Pharmacopoeia Convention Rockville, MD.
(27) GAMP Good Practice Guide A Risk Based Approach to GXP Compliant Laboratory Computerised Systems, Second Edition 2012, Tampa, FL: International Society for Pharmaceutical Engineering.
(28) R.D. McDowall, Spectroscopy32(9), 24–30 (2017)
(29) P.E. Smith and R.D. McDowall, LCGC Europe 31(7), 385–389 (2018).
(30) P.E. Smith and R.D. McDowall, LCGC Europe31(9), 504–511 (2018).
(31) R.D. McDowall and C. Burgess, LCGC North Am.33(8), 554–557 (2015).
(32) R.D. McDowall and C. Burgess, LCGC North Am.33(10), 782–785 (2015).
(33) R.D. McDowall and C. Burgess, LCGC North Am.33(12), 914–917 (2015).
(34) R.D. McDowall and C. Burgess, LCGC North Am.34(2), 144–149 (2016).
(35) 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products. 2008, Food and Drug Administration: Sliver Springs, MD.
(36) ICH Q2(R1) Validation of Analytical Procedures: Text and Methodology. 2005, International Conference on Harmonisation: Geneva.
(37) FDA Draft Guidance for Industry: Analytical Procedures and Methods Validation 2000, Food and Drug Administration: Rockville, MD.
(38) FDA Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics. 2015, Food and Drug Administration Silver Springs, MD.
(39) G.P. Martin et al., Pharmacopoeial Forum38(1), 2012.
(40) Concept Paper: Analytical Procedure Development and Revision of ICH Q2(R1) Analytical Validation. 2018, International Council on Harmonisation: Geneva.
R.D. McDowall is the director of RD McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com








: An Interview With Fabrice Gritti")




, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.


Using GC-MS to Measure Improvement Efforts to TNT-Contaminated Soil
April 29th 2025Researchers developing a plant microbial consortium that can repair in-situ high concentration TNT (1434 mg/kg) contaminated soil, as well as overcome the limitations of previous studies that only focused on simulated pollution, used untargeted metabolone gas chromatography-mass spectrometry (GC-MS) to measure their success.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

Potential Obstacles in Chromatographic Analyses Distinguishing Marijuana from Hemp
April 28th 2025LCGC International's April series for National Cannabis Awareness Month concludes with a discussion with Walter B. Wilson from the National Institute of Standard and Technology’s (NIST’s) Chemical Sciences Division regarding recent research his team conducted investigating chromatographic interferences that can potentially inflate the levels of Δ9-THC in Cannabis sativa plant samples, and possible solutions to avoid this problem.

