Current Good Manufacturing Practice (cGMP): An Overview for the Analytical Chemist
Good Manufacturing Practice (GMP) is the most significant regulation impacting the pharmaceutical industry and requires substantial investment in resources and time commitments. This paper provides a high-level overview of Current Good Manufacturing Practice (cGMP) regulations and related public quality standards. It discusses GMP compliance requirements and practices, emphasizing those relevant to the analytical chemist in development and quality control.
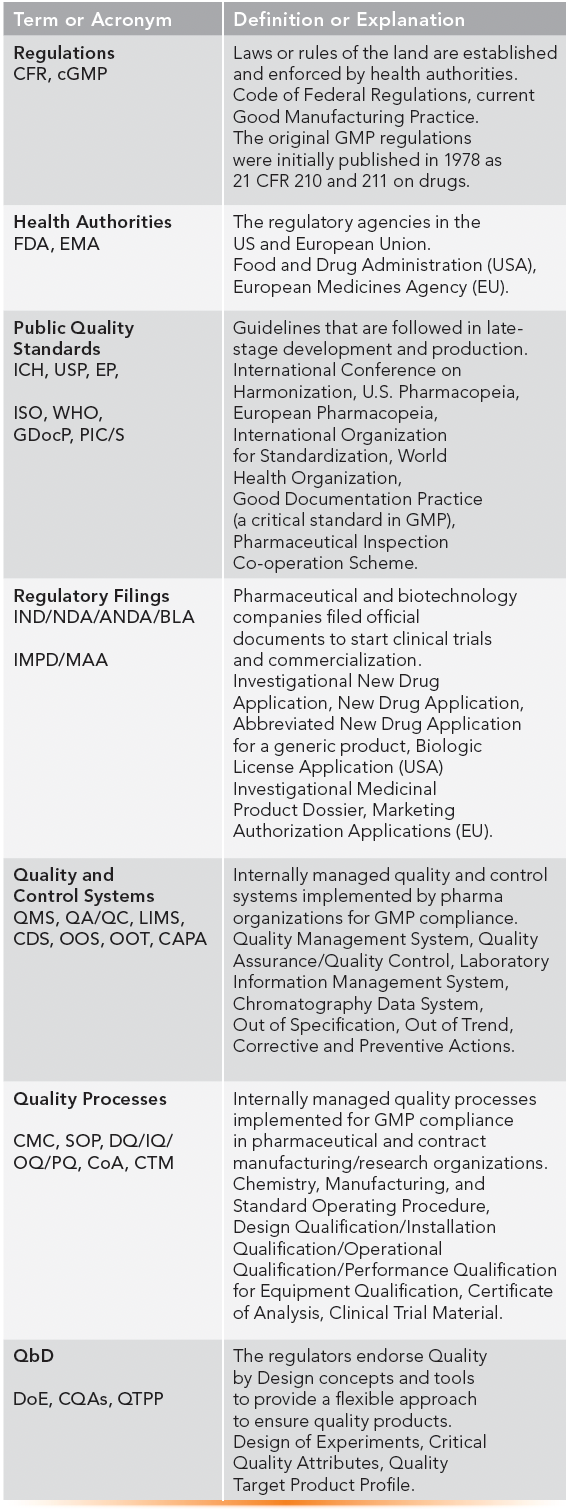
The original cGMP regulations published in 1978 in Codes of Federal Regulations (CFR), 21 CFR 210 and 211 (1,2), contain requirements for methods, facilities, and controls for manufacturing, processing, packaging, and testing drug substances and products. The regulations are designed to ensure that a product has the safety, identity, purity, potency, and clinical efficacy it claims to have. 21 CFR 210 is a brief introductory document that provides a general overview of the minimum requirements of GMP in manufacturing, processing, packaging, or holding of drugs, including status, applicability, and definitions of terms. 21 CFR 211 is a longer document that describes the requirements for finished pharmaceuticals and contains eleven subparts from A to K (A. General provisions; B. Organization and personnel; C. Building and facilities; D. Equipment; E. Control of components and drug product containers and closures; F. Production and process controls; G. Packaging and labeling control; H. Holding and distribution; I. Laboratory controls; J. Records and reports; and K. Returned and salvaged drug products). Since technology is continuously advancing, compliance with the most current standards is expected. Thus, the term “current” in cGMP is used. Table I lists the terms and acronyms used in the paper.


TABLE I: Glossary of terms and abbreviations
GMP regulations provide guidelines and minimum requirements in manufacturing and development practices and facilities to ensure products are consistently safe, pure, and effective for their intended use (1,2). Although the original regulations (21 CFR 210 and 211) are for small-molecule drug products (1–3), GMP compliance is also required for biologics (4), some foods (5), medical devices for human use (6), dietary supplements (7), and other critical quality processes (8,9). Table II lists the U.S. GMP regulations established, amended, and enforced by the US FDA. Other associated regulations from the EMA, public quality standards, and guidelines from ICH, WHO, ISO, and USP were described in our previous paper #4 (10), and they are discussed further here.

TABLE II: A list of cGMP regulations in the United States
The complex and multifaceted GMP regulations and compliance requirements and practices can be found in the regulatory source documents (1–9) and public guidance (10,11), textbooks (12–15), book chapters (16), journal review articles (17–19), and training resources from university programs (for example, regulatory affairs or pharmaceutical sciences) (20) and professional training courses (21). These resources and the company’s quality systems or SOPs should be referred to for definitive guidance in GMP activities.
This paper aims to provide a high-level overview of the cGMP regulations and their principles, intents, and compliance practices. This article is the sixth in a series of nine white papers on “Pharmaceutical Industry for the Analytical Chemist,” to be published in LCGC from 2022 to 2024. Paper #4 provides an introductory overview of pharmaceutical regulations, public quality standards, and overall compliance processes (10). Paper #5 describes Good Laboratory Practice (GLP) for early-phase nonclinical safety, pharmacokinetic studies, and bioanalytical assays (22).
GMP Compliance: From Regulations to Internal Quality Processes
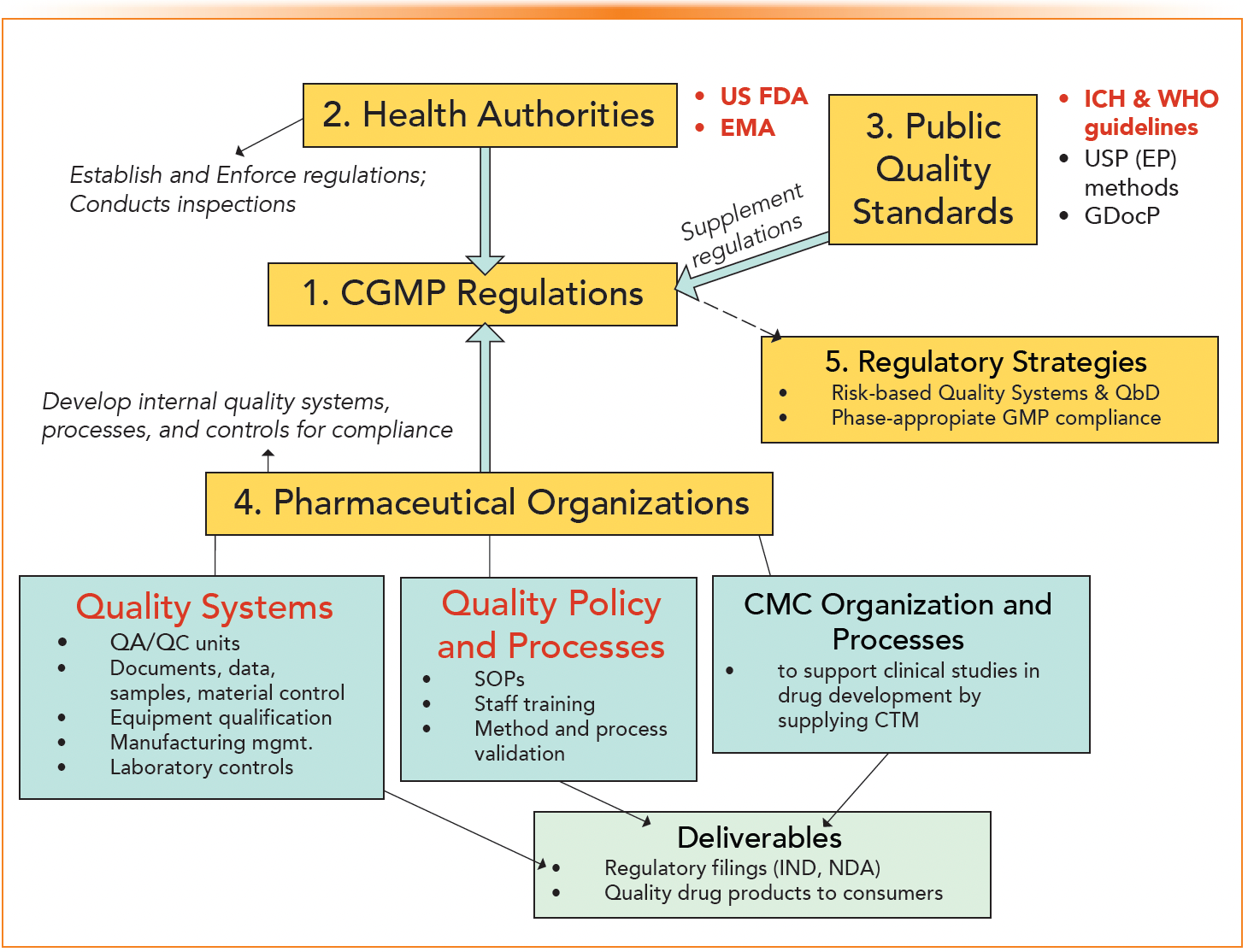
Since health authorities (regulatory agencies) only address the minimum GMP requirements in the Code of Federal Regulations without providing details on how to comply, the company must understand the regulations for its product category and devise its own quality systems and processes for compliance. To help the reader in understanding these complex and interactive processes, we categorized the compliance scenario into five sections (10):
- Pharmaceutical regulations (law of the land promulgated by legislation),
- Health authorities (establish and enforce the regulations),
- Public quality standards (supplement the regulations in scientific terms),
- Pharmaceutical organizations (establish internal quality systems and procedures),
- Regulatory trends and approaches (Quality by Design [QbD], risk-based quality systems, and stage-appropriate GMP compliance in drug development).
Figure 1 is a pictorial portrayal of the five categories extracted from reference 10 and revised for GMP regulations.

FIGURE 1: A pictorial portrayal of the pharmaceutical regulatory processes, categorized into five sections to help readers better understand these complex compliance processes.
GMP Regulations in Non-US Countries
GMP Regulations from the European Commission
For companies having manufacturing sites in the European Union, EU GMP regulations are listed in the EudraLex guidelines (23), which have eleven sections with some similarities to the US counterparts (1,2).
- Scope
- Principles
- Pharmaceutical Quality System (PQS)
- Premises
- Equipment
- Utilities
- Personnel
- Production and specific technologies
- Environmental and process monitoring
- Quality control (QC)
- Glossary
World Health Organization (WHO) Guidance for Other Countries
Countries or economic regions that do not have their own GMP regulations often reference the WHO guidelines (24). WHO provides numerous guidance documents spanning development through production (25). Considering the diversity of global regulations, the Pharmaceutical Inspection Co-operation Scheme (PIC/S) was established in 1995 and is a non-binding, informal cooperative arrangement between more than 50 regulatory authorities in the field of GMP for medicinal products for human and veterinary use. PIC/S is the sole organization that exclusively deals with GMP and provides harmonized standards and training for inspectors worldwide (26).
Although PIC/S is not a trade agreement, a variety of mutual recognition agreements (MRA) exist between major regulatory agencies such as the FDA, EMA, Health Canada, PMDA, and others. The goals of MRAs include:
- Relying on each other’s GMP inspection systems
- Sharing information on inspections and quality defects
- Waiving batch testing of products on import into their territories.
MRAs are typically narrow in scope, for example, excluding vaccines or plasma-derived products. One example of an MRA is that the EU Member States will not duplicate inspections conducted by the FDA for human medicines (27).
Public Quality Standards (ICH Guidelines and USP Methods)
Augmenting the regulations are public quality standards or guidelines to which pharmaceutical companies generally adhere even though they are not considered regulations. Our overview (10) describes these quality standards and organizations, and we will comment further on ICH guidelines and USP compendial methods.
International Conference on Harmonization (ICH guidelines)
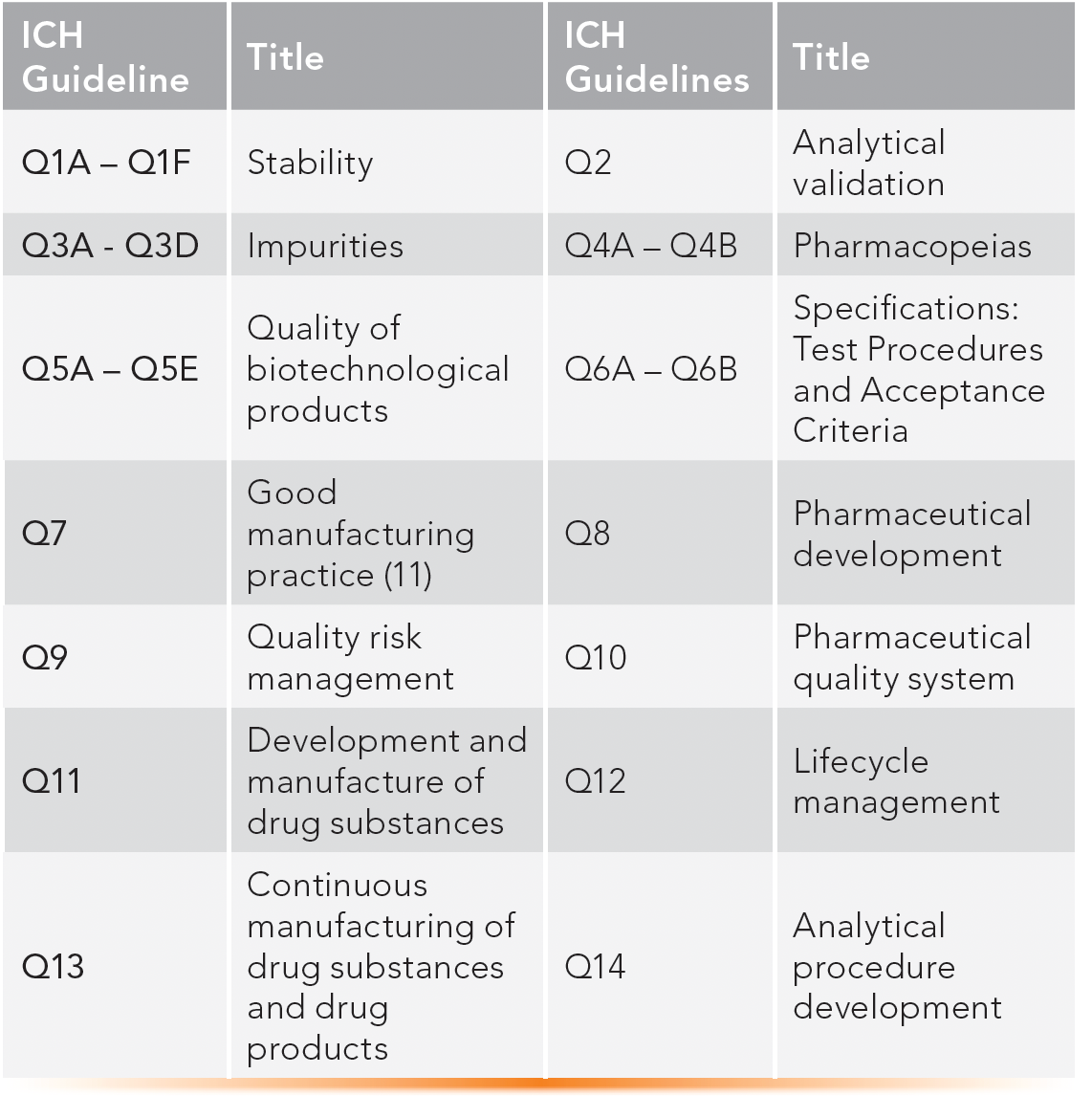
ICH brings together regulatory authorities of Europe, Japan, and the United States, as well as pharma experts, to discuss scientific and technical aspects of product registration. ICH issues guidance documents in Quality, Safety, Efficacy, and Multidisciplinary topics (QSEM) (28). For analytical development and quality control chemists working under GMP, the most referenced documents are the ICH quality guidelines (Q guidelines) listed in Table III.

TABLE III: ICH quality guidelines commonly used in drug development and production.
United States Pharmacopeia (USP)
USP is an independent, nonprofit organization whose mission is to improve world health by setting public standards to help ensure the quality, safety, and benefits of medicines and foods (29). They issue the USP-NF, a combination of two compendia, the United States Pharmacopeia and the National Formulary.
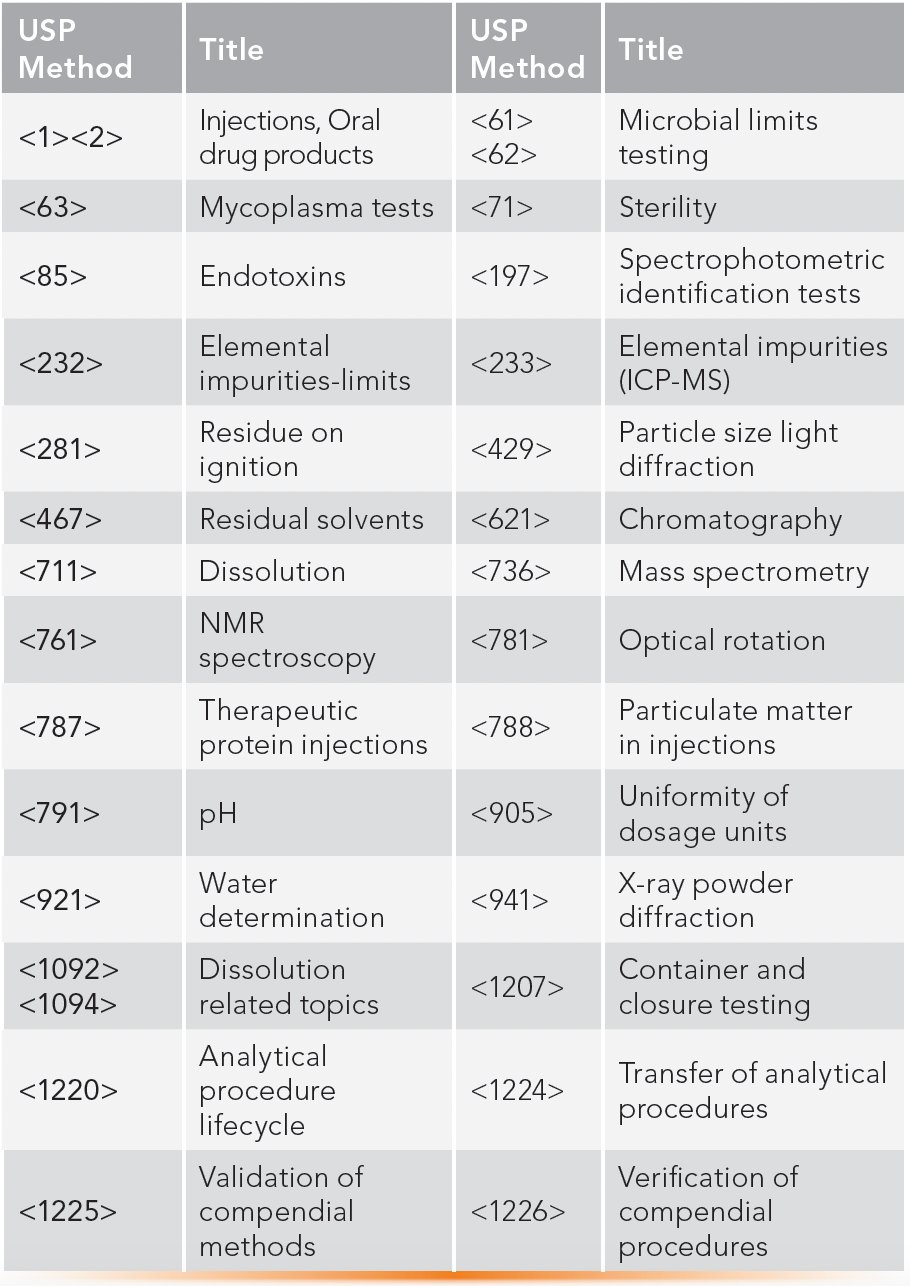
USP monographs contain tests, procedures, and acceptance criteria frequently employed to facilitate consistency in testing across the generic industry. Monograph testing requires the use of compendial reference standards. The USP also contains general chapters on standard techniques and general methodologies applied across many monographs used in specifications and certificates of analysis (CoA) (16–18). Also, the general chapters cover a broader scope, like method validation, verification, and method transfer. Validation of compendial methods is not required but must be verified for suitability for the intended use of the sample in your laboratory according to USP <1226>. The common USP methods often used in specifications and CoAs are listed in Table IV.

TABLE IV: Listing of common USP compendial methods in drug analysis
GMP Violations and Enforcement Actions by US FDA
If a company is not complying with cGMP, any drug it makes is considered “adulterated” under the law. This kind of adulteration means that the drug was not manufactured under conditions that comply with cGMP. Regulatory actions against companies with unacceptable cGMP compliance are intended to prevent the possibility of unsafe and ineffective drugs from getting to market (10,30). Observations found during inspections (GMP and pre-approval inspections [PAI]) by the FDA are formally documented using Form 483. Subsequent corrective actions by the company to address 483 observations can be quite involved. Repeated violations may result in the company receiving a warning letter from the FDA. Although the FDA cannot force a company to recall a drug, companies usually will recall voluntarily or at the FDA’s request. The FDA can warn the public and seize the drug in extreme cases. Seizure and injunction cases often lead to a court order and a subsequent “consent decree” that requires companies to take defined corrective steps to correct cGMP violations. Companies may be prohibited from distributing products until the consent decree is lifted (30).
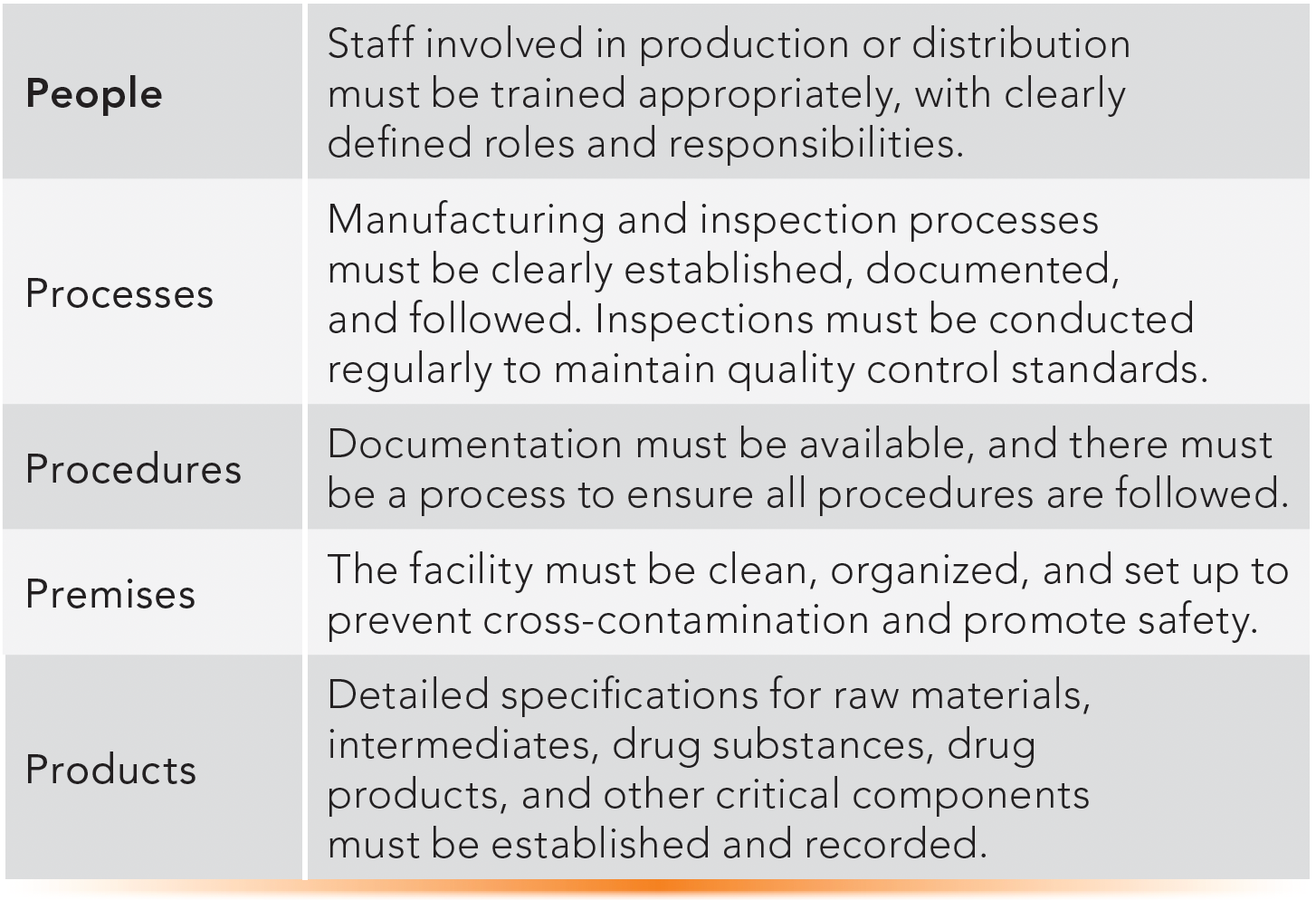
Some Basic GMP Elements: The Five “P’s”
GMP regulations are formal, detailed, and complex. They include elements related to specific operations or business categories, such as manufacturing, process validation, or product recalls. However, some basic principles apply to every operation. Table V describes common elements of cGMP, sometimes called the “5 P’s”—people, processes, procedures, premises, and products (31).

TABLE V: Basic cGMP elements: the five “P’s” in GMP
GMP in Drug Development and QC Laboratories
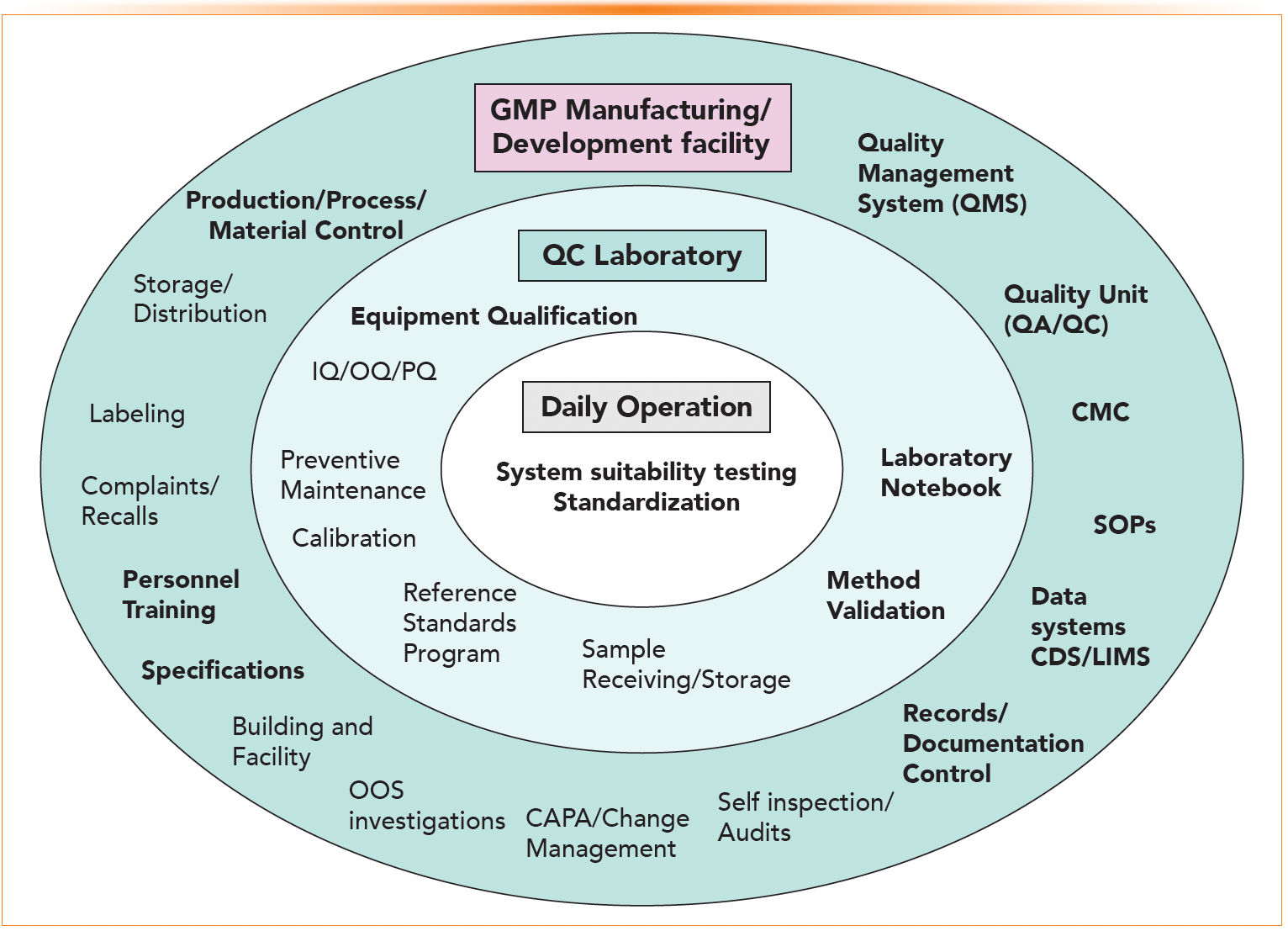
Figure 2 shows an example of the organizational structure of quality systems and processes in a GMP manufacturing/ development facility, including those within a QC laboratory. Figure 2 was extracted from reference 10 and revised to add further components for GMP.

FIGURE 2: An example of the organizational structure of quality systems and processes in a GMP manufacturing and development facility, including within a QC laboratory. This figure is extracted from reference 10 and revised to include additional GMP components. Bold texts are components of more relevance in the workflow of QC analysts.
Quality by Design (QbD)
Regulatory agencies encourage companies to adopt a Quality by Design (QbD) approach to drug development (32), such that quality is designed into the entire manufacturing process rather than relying solely on QC testing at release. This is achieved by gaining product and process knowledge during development. Organizations will define a Quality Target Product Profile (QTPP) for the drug and use risk assessment techniques to identify Critical Quality Attributes (CQAs) to be monitored during production and at release. The analytical development chemist will then develop and validate methods using a multivariate DoE approach to execution and analysis to assess CQAs captured in product specifications for release testing (16,19,33–34). QbD also utilizes a lifecycle approach or continuous method improvement throughout the product lifecycle. For example, the method will be monitored over time to identify trends such as system suitability failures or unusually high numbers of deviations. The analytical chemist may redevelop and revalidate the method because of changes in process (such as for quantitation of new degradation products) or for method improvement purposes.
Phase-Appropriate GMP Compliance Approach
As described elsewhere (10,14,17), regulatory oversights increase throughout drug development. In late-phase development laboratories, analytical chemists develop and validate robust methods suitable for testing registration stability lots, releasing clinical trial materials (CTM) and transferring to manufacturing sites within the company or at a third party. Method validation attributes are described in ICH Q2 (R2), USP <621>, <1225>, and other references (16,19). Typical validation attributes include specificity, accuracy, linearity, range, precision (repeatability and intermediate precision), limits of detection and quantitation, robustness, and system suitability (including stability of standard and sample preparations) (19). As these late-phase procedures are intended to be transferred to QC units in the manufacturing facilities of commercial batches, procedures and records should be sufficiently detailed to ensure the transfer will be successful. Guidance for analytical method transfer studies is given in USP <1224>. Equipment in these labs must be fully qualified (IQ/OQ/ PQ) (16), and computerized systems must comply with 21 CFR Part 11 requirements for electronic records and signatures (8). This provides assurance the equipment is performing as intended and that data cannot be manipulated or erased.
Good Documentation Practice (GDocP)
There is no specific regulation on Good Documentation Practice (GDocP), though it is a central pillar of GMP compliance. GDocP includes the principles of “ALCOA+,” which stands for Attributable, Legible, Contemporaneous, Original, Accurate (plus Complete, Consistent, and Enduring) (35). These are integral to cGMP processes, as the main objective of reliable and traceable documentation is to establish, control, monitor, and record all activities that impact product quality. The term GDP is often used, though it should not be confused with Good Distribution Practices for manufacturing facilities.
The QC Analyst
A QC analyst is a laboratory scientist whose primary duties are to measure and test raw materials, active ingredients, in-process control and intermediates, excipients, and finished products at a manufacturing facility, contract manufacturing organization (CMO), or contract research organization (CRO). The QC analyst may be responsible for stability testing and participating in investigations of out-of-trend or out-of-specification results. Testing is controlled through general SOPs, work instructions, and analyte-specific test methods. The test method must be validated in a GMP laboratory using 21 CFR Part 11 compliant data systems (16,18). For example, OOS results and deviations are reported, investigated, and recorded. Their work is subject to inspection and audit since the analytical results they generate determine whether raw materials are dispositioned for use in compounding drug products or whether drug products comply with specifications for release and distribution to patients. Deficiencies found during inspections and audits lead to a CAPA to resolve the deficiency and avoid its recurrence.
The Restrictive GMP Work Environment for QC Analysts
Many laboratory scientists working under GMP are unhappy about the restrictive work environment in adhering to numerous SOPs and time-consuming documentation and review processes. In an ideal world, a quality system should be possible with efficient procedures for GMP compliance. Nevertheless, after reacting to deviations from audits and inspections, many companies’ quality systems and procedures have grown to be overly complex and bureaucratic with time. These inefficient procedures burden the daily routines of the QC analyst and prolong the overall drug development timeline. Pharmaceutical companies may benefit from a holistic re-evaluation of their internal policies and processes to identify opportunities to streamline workflows without impacting compliance.
Conclusions and Summary
cGMP regulations provide minimum quality standards to ensure quality is built into pharmaceuticals’ design and manufacturing process. The analytical development and QC analyst are essential to compliance by adhering to cGMP guidelines. Hence, their data are valid, traceable, and reproducible, ensuring that products are safe and efficacious.
Disclaimer
This paper provides a high-level overview of cGMP regulations and related public quality standards for pharmaceutical scientists. Although the goals of GMP are straightforward, compliance activities are diversified and variable, driven by the company’s SOPs, resources, and compliance culture. The information presented here stems from health regulations, guidance documents, references, and the authors’ experience. Presenting a concise overview of this complex topic is challenging. The reader should refer to the original regulations and guidelines, references, and internal SOPs for more definitive compliance activities in the laboratory.
Acknowledgments
The authors thank the reviewers for providing timely technical and editorial inputs to this article: Alice Krumenaker of Hovione, Anissa Wong of Gilead, David VanMeter of Sartorius, He Meng of Sanofi, Mike Shifflet of Kenvue, Kate Evans from Longboard Scientific, and Zedong Dong of Innovent Biologics.
References
(1) CFR Title 21 - Food and Drugs, Part 210 - Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs, Government Publishing Office.
(2) CFR Title 21 CFR Part 211 - Good Manufacturing Practice for Finished Pharmaceuticals. Government Publishing Office:
(3) CFR Title 21, Part 212, Current Good Manufacturing Practice for Positron Emission Tomography Drugs, Government Publishing Office.
(4) CFR Title 21, Parts 600 and 610, Good Manufacturing Practice Biologics, Government Publishing Office.
(5) CFR Title 21, Part 110, Good Manufacturing Practice for Human Food, Government Publishing Office.
(6) CFR Title 21, Part 820, Quality System Regulation (Medical Devices), Government Publishing Office.
(7) CFR Title 21, Part 111, Good Manufacturing Practice for Dietary Supplements, Government Publishing Office.
(8) CFR Title 21, Part 11, Electronic Records; Electronic Signatures, Government Publishing Office.
(9) CFR Title 21, Part 314, Application for FDA Approval to Market a New Drug, Government Publishing Office.
(10) Doneski, L., Dong, M. W. Pharmaceutical Regulations: An Overview for the Analytical Chemist, LCGC North Am. 2023, 41 (6), 211–215. DOI: 10.56530/lcgc.na.ua3181v7
(11) ICH Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients, US FDA, 2016.
(12) Durivage, M. A. (Ed.) The Certified Pharmaceutical GMP Professional Handbook, 2nd Ed., ASQ Quality Press, 2016.
(13) Nally, J. D., Ed. Good Manufacturing Practices for Pharmaceuticals 6th Ed., CRC Press, 2007.
(14) McBride, L.; Schmitt, S. (Eds.) Fundamentals of Pharmaceutical and Biologics Regulations: A Global Perspective, Regulatory Affairs Professionals Soc. 2023.
(15) Huynh-Ba, K. (Ed.), Analytical Testing for the Pharmaceutical GMP Laboratory, Wiley, 2022.
(16) Dong, M. W. HPLC and UHPLC for Practicing Scientists, 2nd Ed., Wiley, 2019. Chap. 9 and 11.
(17) Dong, M. W. Drug Development Process: Nonclinical Development of Small Molecule Drugs, LCGC North Am. 2022, 40 (10), 484–492. DOI: 10.56530/lcgc.na.lz5690w8
(18) Kou, D.; Wigman, L.; Yehl, P.; Dong, M. W. Separation Science in Drug Development, Part 4: Quality Control, LCGC North Am. 2015, 33 (12), 900–909.
(19) Dong, M. W., Huynh-Ba, K., Wong, A. W. Validation of Stability-Indicating HPLC Methods for Pharmaceuticals: Overview, Methodologies and Case Studies, LCGC North Am. 2020, 38 (11), 606–618.
(20) Singh, N., Good Manufacturing Practices, online course at U. C. Santa Cruz Silicon Valley, Extension, 2024.
(21) Dong, M. W., Drug Quality Fundamentals, Quality Control of Small Molecule Drugs and Recombinant Biologics, Course at U. C. Santa Cruz Silicon Valley Extension, 2017.
(22) Doneski, L.; Roos, D.; Dong, M. W. Good Laboratory Practice: An Overview for the Analytical Chemist, LCGC North Am. 2023, 41 (9), 381–385. DOI: 10.56530/lcgc.na.un1878g5
(23) EudraLex - Volume 4 - Good Manufacturing Practice (GMP) guidelines, European Commission, 2022; https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en (accessed 2023-10-16)
(24) World Health Organization, TRS 1044 - Annex 6: WHO good practices for research and development facilities of pharmaceutical products, WHO Technical Report Series, WHO, 2022.
(25) Gaur, A., International Harmonization via ICH, WHO, and other Global Initiatives in Fundamentals of Pharmaceutical and Biologics Regulations: A Global Perspective, Regulatory Affairs Professionals Soc. 2023, pp 23.
(26) Pharmaceutical Inspection Co-operation Scheme (PIC/S), PE-009-17 (Parts I & II), Pharmaceutical Inspection Convention, 2023.
(27) Questions & Answers on the impact of Mutual Recognition Agreement (MRA) between the European Union and the United States as of 31 May 2023, EMA, https://www.ema.europa.eu/en/documents/other/questions-answers-impact-mutual-recognition-agreement-between-european-union-united-states-31-may_en.pdf (accessed 2023-10-16)
(28) International Conference on Harmonization, https://www.ich.org/page/quality-guidelines (accessed 2023-10-16)
(29) United States Pharmacopeia (USP), https://www.usp.org/ (accessed 2023-10-16)
(30) Facts About the Current Good Manufacturing Practices (CGMP), US FDA, https://www.fda.gov/drugs/pharmaceutical-quality-resources/facts-about-current-good-manufacturing-practices-cgmp (accessed 2023-10-16)
(31) GxP: The 5 Ps of Good Practice, Manufacturing Chemist, 2019, https://www.manufacturingchemist.com/news/article_page/GxP_The_5_Ps_of_Good_Practice/153485 (accessed 2023-10-16)
(32) Nasr, M. M. Implementation of Quality by Design (QbD): Status, Challenges and Next Steps, CDER, FDA, 2006.
(33) Validation of Analytical Procedures: Text and Methodology Q2(R1), ICH, 2005.
(34) Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics, FDA, 2015.
(35) TRS 966 - Annex 5: WHO good data and record management practices.
ABOUT THE CO-AUTHORS

Leon Doneski has over 20 years of experience in the pharmaceutical industry, contributing to many product registrations. He is currently the Director of Regulatory CMC for Arcutis Biotherapeutics. Leon earned his PhD in analytical chemistry from the University of Delaware.

Michael W. Dong is a principal of MWD Consulting, which provides training and consulting services in HPLC and UHPLC, method improvement, pharmaceutical analysis, and drug quality. He was formerly a Senior Scientist at Genentech, a Research Fellow at Purdue Pharma, and a Senior Staff Scientist at Applied Biosystems/ PerkinElmer. He holds a PhD in Analytical Chemistry from City University of New York. He has more than 130 publications and a best selling book in chromatography. He is an editorial advisory board member of LCGC North America and the Chinese American Chromatography Association. Direct correspondence to: LCGCedit@mmhgroup.com.















Characterizing Polyamides Using Reversed-Phase Liquid Chromatography
May 5th 2025Polyamides can be difficult to characterize, despite their use in various aspects of everyday life. Vrije Universiteit Amsterdam researchers hoped to address this using a reversed-phase liquid chromatography (RPLC)-based approach.



New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

