COVID-19 and Data Integrity: What Does Lockdown Mean for Chromatograph Qualification and Calibration?
LCGC North America
How should you maintain the calibration and qualification of chromatographs during the COVID-19 pandemic? Here, we discuss the details of how to ensure data integrity of results when faced with travel limitations and requirements for social distancing.
The manufacture of pharmaceutical products is classed as critical work, and thus is continuing during the COVID-19 pandemic. With limitations of travel and requirements for social distancing, how should you maintain the calibration and qualification of chromatographs to ensure data integrity of results during this pandemic?
Over the past three months, as coronavirus or COVID-19 has spread from China around the world, many counties have engaged in various forms of lockdown, social distancing, or travel restrictions to prevent the spread of the virus. The focus in the news has naturally been on the number of infections identified, the number of patients in hospitals, and, sadly, on the number of deaths. In the background, the pharmaceutical and medical device industries are still working to produce medicines such as acetaminophen (paracetamol) to alleviate the fever caused by the virus, developing potential vaccines, and conducting research to see if any existing medicines can combat the disease.
During this pandemic, it can be easy to forget that we are a regulated industry, and, in the laboratory, chromatographs must be qualified, calibrated, and maintained. To ensure that this is the case, inspections are carried out to ensure that laboratories are complying with regulatory requirements. However, the direct impact of the pandemic on regulated laboratories is an important subject that needs to be considered. In some instances, facilities are restricting access to sites to try to minimize potential risk of exposure for critical employees, while, in others, restrictions may prevent service engineers from traveling. Both issues have the potential to affect compliance in a laboratory. In the absence of inspections and service engineers, how will you keep your chromatographs operating reliably and ensuring data integrity?
Why Are Qualification and Calibration Important to Data Integrity?
A data integrity model has been presented and discussed in an earlier “Data Integrity Focus” article (1), and also in my book, Data Integrity and Data Governance (2).
The model consists of four layers. The foundation layers consist of management leadership, quality culture and ethics, procedures, and training. Level 1 requires the right analytical instrument for the job. The importance of Level 1 to data integrity is if you don’t know how an instrument performs, how can you trust the results it generates? Therefore, at Level 1, analytical instruments must be qualified under USP chapter <1058> on Analytical Instrument Qualification (3), and comply with the requirements of 21 CFR 211.63 and 160[b](4) (4) and EU GMP Chapter 3 clauses 3.34 and 3.35 (5). As the FDA issued 17 (211.63) and 37 (160[b][4]) 483 observations against these two CFR codes in 2019, these areas are on an inspector’s radar (6). We discuss two of these regulations later in this article.
What Happens Now? Inspections
Let us have a look at what is happening with two regulatory agencies: the Food and Drug Administration (FDA) in the United States, and the Medicines and Healthcare Products Regulatory Agency (MHRA) in the United Kingdom.
In a press statement issued on March 10th of this year, the FDA announced that they were postponing all foreign inspections due to travel advice from the State Department that prohibits foreign travel by U.S. government employees. In addition, there are actions by non-U.S. governments that require overseas travelers to self-isolate for 14 days on arrival, or prohibit any foreign travelers arriving at all. Therefore, given that the FDA are unable to physically inspect foreign companies, they will gather information under Mutual Recognition Agreements with other regulatory authorities, and revert to physical inspection and possible testing at the U.S. border, as they have done since the start of the outbreak in China when inspections were halted there (7).
Later, the FDA announced on March 18th that domestic routine surveillance inspections were being postponed for the safety of the inspectors, and also due to industry concerns about visitors. However, for-cause inspections would be risk assessed, and only critical ones would be carried out (8).
In the United Kingdom, the MHRA posted a statement on the web site on March 20th, stating that they are only conducting essential good practice (GxP) inspections associated with the UK’s response to COVID-19, and all other inspections will be deferred to a later date (9). The same post noted that:
We are expecting organizations to maintain GxP compliance, and will support the industry and NHS to focus on service continuity by using alternative approaches for routine regulatory oversight, such as office-based assessment and the sharing of information within the international regulatory network.
On March 30th, there was explicit advice posted with specific guidance for GLP studies (10):
There will be office based inspections.
Still important to maintain compliance with principles of GLP.
David Churchward expanded the overall approach in an MHRA blog explaining that inspectors were working from home as much as possible. Office-based inspections consisted of a request of documents from a company by e-mail, and, after review, an inspector would follow up with either e-mail questions or a teleconference (11).
Although you may not see an inspector for a time, which in some circles may be a cause for celebration, there is still the regulatory expectation that you remain in compliance. So that is all well and good.
However, consider what happens if you have a chromatograph that is due for a preventative maintenance (PM) visit and annual operational qualification (OQ), and the engineer can’t get to you, or your company won’t let them come on-site. Grace periods for instruments typically only last for one to four weeks. Let’s look closer at the details of two regulations to help us.
Regulations for Qualification and Calibration
The first of the key regulations is 21 CFR 211.160(b)(4):
The calibration of instruments, apparatus, gauges, and recording devices at suitable intervals in accordance with an established written program containing specific directions, schedules, limits for accuracy and precision, and provisions for remedial action in the event accuracy and/or precision limits are not met. Instruments, apparatus, gauges, and recording devices not meeting established specifications shall not be used (4).This means that the specifications of the chromatograph should be documented in a user requirement specification (URS) or equivalent under GMP, as well as required by USP <1058> (3).
Also pertinent is EU GMP Chapter 3, clause 3.35:
Repair and maintenance operations should not present any hazard to the quality of the products (5).
If any repairs or maintenance are made to a chromatograph, then it must be partially or fully requalified to demonstrate fitness for purpose for analysis during the production of investigational medicinal products (IMPs) or pharmaceutical products.
What Happens to Service and Qualification in Lockdown?
Apocryphally, the way companies perform qualification and maintenance breaks into two groups. Approximately two thirds of companies will use a third party to service, maintain, and qualify their analytical instruments. Larger sites with a lot of instrumentation from a single supplier, or, more likely with a service provider able to service, maintain, and qualify a range of instruments from different suppliers, may have a service engineer stationed permanently on-site. In this case, the engineer is known, and may be treated differently from one who travels to the site. The remainder of companies (approximately one third) use a combination of in-house engineering and metrology groups to service and qualify their instruments, and only use the original supplier if there are major breakdowns (for example, if the instrument cannot be used).
The problem with the inspectors now comes to service engineers. Are they counted as essential workers when a state or country is in lockdown? Even if they could travel, would an organization allow them on-site, where they could potentially infect staff? Normally, companies may only consider risk to the patient from the impact of errors, mistakes, or problems (which is covered later in this article). In this situation, batches are not released where there is unacceptable risk. However, in this pandemic, failure to supply may represent a greater risk to the patient, than the risks identified if instruments are used with limited compliance. For example, in March 2020, the FDA issued guidance for industry reminding companies of the requirement to notify the FDA of permanent discontinuance or interruption in manufacturing (12). Failure to do so could result in the FDA issuing a noncompliance letter. While failure to supply is a risk to patient, how should firms balance the potential risks of noncompliance? Where does ethics come into this equation?
Consider what happens if you have a chromatograph that requires a PM followed by an OQ now.
Balancing and Managing Risks
We must consider a number of risks, and the management of them, in this unusual situation.
Typically, a laboratory has multiple chromatographs at its disposal. If the PM/OQ dates of the instruments are spread throughout the year, it might be possible to switch from one instrument to another to mitigate the risk associated with the lack of maintenance and qualification. However, some organizations may synchronize their maintenance and qualification visits over a short period of time, to save on an engineer’s travel time and costs. In this situation, all chromatographs can be out of maintenance and qualification at virtually the same time.
However, if the lockdown continues, it is likely that the dates of maintenance and qualification of all chromatographs will become overdue. Understand that, at the expiry of a PM or OQ due date and any associated grace period, it does not necessarily mean that a qualification pixie waves a magic wand and all chromatographs are no longer calibrated or qualified and they must not be used. However, there is a deviation from the maintenance and qualification schedule that must be documented.
If chromatographs are dedicated to specific methods, one way to increase flexibility could be to use an instrument for any analysis. If this is the case, a laboratory should check the ranges of qualification performed on the instrument to ensure that they are not exceeded with the new analysis. If this is the case, a laboratory must ensure that the liquid chromatography (LC) system is thoroughly washed and flushed through between different analyses, including autosampler needle washes; otherwise, problems may occur.
These gaps and the associated risks must be assessed, evaluated, controlled, and, above all, documented. As part of the risk control strategy, overdue maintenance and qualification might be justified and based on data, such as:
- If an LC instrument is not used frequently, it might not require maintenance even if overdue.
- The total number of sequences and injections might increase or decrease the overall risk.
- The type of therapeutic product being analyzed on the instruments in question (and the product’s therapeutic range, range of individual parameters measured, route of administration, age of patients treated, and so on) might also have an impact on the severity of an over- or understrength product being released to the market (if the instrument used to ensure the products’ quality were out of calibration at the time of analysis).
Trending of test results, as required by EU GMP Chapter 6.9 (13), might also support any risk acceptance.
The risks associated with any action are dependent on the context from which the actions are considered. So, in this instance, the risks to the patient associated with failure to supply pharmaceuticals must be balanced against the risks that result from instruments that cannot be fully maintained or qualified. If the instruments are not in use, and will remain so, the only risks are the possible noncompliance with procedures, such as grace periods associated with maintenance, and qualification and consideration of how to return the instruments to use in a compliant way after the period of disruption. It is probable that, in the post COVID-19 period, the demand for service personnel will spike and will exceed capability. Companies with a service contract will be prioritized over those that request on-demand services. If your laboratory falls into the latter category, be
prepared for a long wait for service and requalification.
If a decision is made to continue using instruments outside of maintenance and qualification grace periods, the principal types of risk associated with this action include:
- Breakdown: Instruments not maintained are more likely to break down during use.
- Regulatory: Justification of actions when the next inspection occurs. This can include defending the decision to continue using the instruments, and documenting scientifically that using instruments without maintenance and full qualification were under control and did not impact
- analytical results.
- Patient: Potential risk that substandard or over-strength pharmaceutical may be released.
Any decision to use an instrument with overdue maintenance or qualification must be documented carefully, and in a transparent manner. The worst possible way to document a sensitive decision such as this would be to try to hide it. Historically, companies may have issued a miniature risk assessment or file note that documented sensitive decisions, but hiding where the document is buried with no traceability to it within corporate quality systems. Such action would now be interpreted as running a secondary quality system from a data integrity perspective (14).
What Can Go Wrong with an HPLC Instrument?
If your laboratory cannot obtain service, and you decide to continue using your chromatographs, you must perform an enhanced risk assessment to support this decision. You need to try to identify problems with a chromatograph before and during any analysis. In 2015, Paul Smith and I published a “Questions of Quality” article in LCGC Europe that is the basis of this section about risk assessment of a high performance liquid chromatography (HPLC) instrument (15). The starting point of our discussion was what can go wrong with an operational HPLC system. The risk assessment article was written from an HPLC perspective, but the principles outlined here (such as, for example, considering how an instrument may fail, and taking appropriate actions to minimize the potential risk associated with the failure) apply equally to other instruments. Because failure modes can be different between different analytical technologies, it is likely that risk assessments for each analytical technique may be needed.
The HPLC risk assessment from 2015 is shown in Figure 1, where a liquid chromatograph consists of four modules: the pump, the injector (autosampler), the column oven, and the detector. The column has been omitted from the figure, as the aim is to look at an LC instrument’s qualification rather than method performance. Underneath each module are listed the main failures that could occur. Note that this list is not exhaustive, and some of the failures could be broken down further. However, to keep the discussion simple, the problem is looked at from a high level. Some failures may not happen if the instrument in your laboratory does not have a particular feature. For example, for an isocratic pump there will not be gradient errors, or with a fixed wavelength detector the wrong wavelength will not be used.


Figure 1: The main possible failures for each module of an HPLC instrument (15).
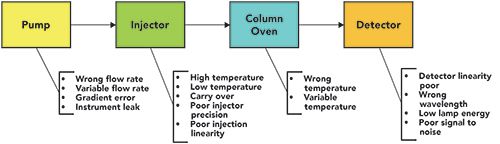
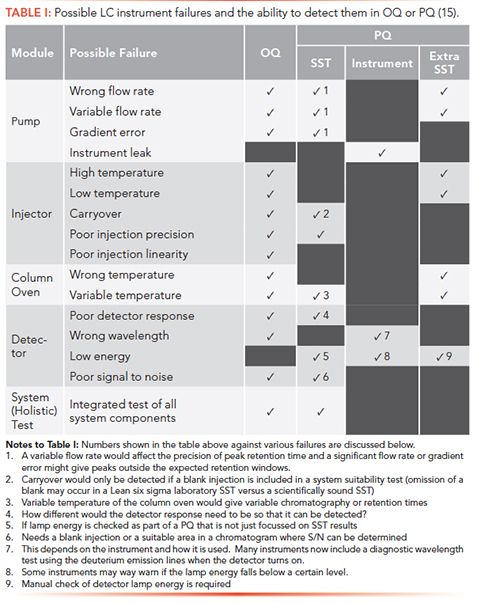
Having listed the main failures, we now need to consider the circumstances under which these might be detected in an OQ or performance qualification (PQ), as shown in Table I. The PQ is broken down into three areas where a failure could be detected: during a system suitability test (SST), by the instrument itself, and during what is called SST extra, where parameters can be measured if the SST is designed to include them, and these can be traced back to the instrument’s user specification.

I’m Overdue for a PM and OQ, But No Engineer is Available
So let’s consider a scenario in which your instrument is overdue for a PM and OQ, and there is no service engineer available for the foreseeable future. Who are you going to call? At this point, the Ghostbusters are probably not a viable option. What are you going to do?
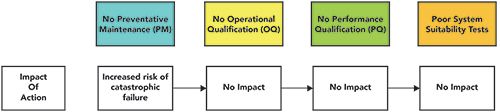
If no PM occurs then and there, one potential impact is an increased risk of a catastrophic failure of the chromatograph, as shown in Figure 2. This is the simplest situation; the instrument becomes inoperative, and there is no further work possible until it has been repaired and requalified. There is, therefore, no impact if there is no OQ, no PQ, or even if there is a poor SST for the method.

Figure 2: Impact of a catastrophic failure of a chromatograph if there is no preventative maintenance.
A more difficult situation arises if there is no PM visit, but the instrument is still operating, because there could be a change in instrument performance over time. Variable performance could result in variable chromatography. Such variability might be detected, but a slow drift in performance that results in method values being outside set points and limits tested during qualification would be much harder to detect, and potentially more serious. As is typical in many laboratories, PM is usually followed by an annual OQ. But in the scenario we are considering, there is no service engineer available, your grace period has expired, probably along with most of the QA department, and no OQ has been performed. This is the situation shown in Figure 3.

Figure 3: The increased risk that performance drift of a chromatograph will not be detected if there is no preventative maintenance, operational qualification, or performance qualification.
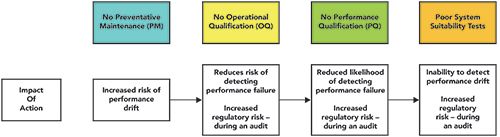
The first issue with no qualification or calibration, per 21 CFR 211.160[b][4], is there is now an immediate regulatory gap with a likelihood of a citation. In addition, given that there is no confirmation that the system is working as intended, there is a risk of not detecting any instrument performance failure. The next fallback would be if the PQ can demonstrate that the instrument meets its intended use as defined in the laboratory URS.
As shown in a recent “Focus on Quality” column that I wrote with Paul Smith, where we discussed PQ for infrared instruments (16), and in the third part of a “Questions of Quality” series on USP <1058> in LCGC Europe for liquid chromatographs (17), PQ is defined by USP <1058> as follows:
PQ is the documented collection of activities necessary to demonstrate that an instrument consistently performs according to the specifications defined by the user, and is appropriate for the intended use.
The PQ verifies the fitness for purpose of the instrument under actual conditions of use. After IQ (instrument qualification) and OQ have been performed, the instrument’s continued suitability for its intended use is demonstrated through continued PQ (3).
You will see, from the definition above, that the PQ is linked back to the URS. Therefore, in the absence of a PM and a current OQ, demonstrating that your instrument still meets its intended use, as defined in the URS, is now your only way for you to check that a chromatograph is still functioning correctly.
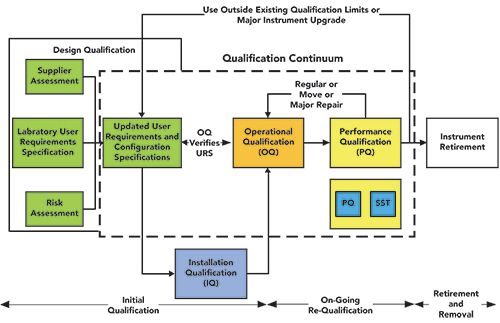
We will come back to discuss this after finding out what is involved in a PQ. The PQ is one part of the analytical instrument qualification (AIQ) continuum that is shown in Figure 4 to demonstrate that the user specifications are being met throughout the use of an instrument.

Figure 4: AIQ continuum from the laboratory user requirements specification (URS) to performance qualification (PQ), adapted from (17).
Under USP <1058> PQ consists of several elements:
- a PQ test plan covering the scope of work to be performed (ideally also risk assessed)
- maintenance activities
- repairs as required
- holistic or method SST with acceptance criteria (3).
Of the items in the list above, only the SSTs can be used to check that the instrument performs as required by the user specifications.
Performing a gap analysis between instrument failure modes and potential detectability of the failure (based on the possible impact on SST) should highlight which failure modes will not be detected with current ways of working.
Performance Qualification Comes to the Fore
In the situation of a missed or delayed OQ due the non-availability of an engineer, the PQ takes on increased significance, as can be seen in Table I and discussed above. Many of the parameters that would be measured directly in the OQ can be indirectly measured in the PQ, as long as the PQ tests applied are related to the laboratory URS, as shown in Figure 4 (17,18). If a laboratory has invested the time and effort in designing appropriate instrument life cycle processes as an integrated AIQ continuum shown in Figure 4, that work will help them remain compliant. For example, if meaningful SST parameters can be linked to the user requirements for the instrument, these companies will be in a stronger position to defend and justify that the potential compliance risks associated with continuing to use instruments outside of grace periods, or even where an OQ is not performed.
Companies that historically perform what might be considered the bare minimum may find themselves facing interesting times when inspections resume. For example, a company that performs an SST measuring only column plate count will find it difficult to provide scientifically sound evidence to support an impact assessment because they simply don’t have appropriate data to support
this decision.
If your approach to SST leans toward the minimum, then, for the period of time the instrument is going to be used outside of grace periods, you should very seriously consider enhancing both chromatography (method) based SST, such as allowed limits on named peaks, and also consider adding non-method-specific point-of-use instrument tests, such as flow rate and temperature measurements.
A Holistic HPLC PQ Test
As part of an overall approach to PQ, there should be a holistic test that can show the user requirements are still being met. Analytical procedures should routinely be designed to be as robust as possible (19). However, the principle of a good holistic PQ test is to design an analytical procedure that is sensitive to instrument performance (which is the opposite of normal analytical science). This is the approach used for performance verification testing (PVT) of dissolution instruments, which is universally interpreted as a PQ for these instruments.
Ideally, the procedure must use stable model compounds, with simple, stable, and robust chromatography to minimize analytical variance from the reference materials or use (17). Standard solutions and mobile phases should be prepared gravimetrically, again to reduce variance, so that instrument problems have a greater probability of being detected. The performance of the procedure is therefore dependent on instrument performance, and can then be related to user requirements, as shown in Figure 4 and Table I. These overall holistic standards allow limits to be set for:
- detector reproducibility and linearity
- autosampler precision
- the combination of the pumping system and thermostatic control of the column.
Unlike an analytical balance, where a calibrated mass set is used to confirm the instrument performance directly with laboratory specifications, this is not possible with a liquid chromatograph. Tests to show that the pump is within specification with a holistic PQ test should ideally be conducted immediately after an OQ, so that the holistic PQ test can be equated with the OQ that can be related directly to the laboratory specification, as shown in Figure 4. Consistency of retention time over a sequence implies that the pump and the oven (if used) are working consistently. All parameters will be measured with the instrument under actual conditions of the chromatograph use, and relate back to
laboratory specifications.
Self Service for Minor Repairs
Minor repairs, such as replacement of pump seals, can be carried out by a laboratory quite easily. However, this requires that these repairs are covered in a standard operating procedure (SOP), and this includes requalification such as measurement of flow reproducibility directly if you have a calibrated flow meter or referencing analytical work such as system suitability criteria recorded to demonstrate the instrument is working appropriately after the repair. During the COVID-19 pandemic, where engineers are not able to come on-site, many service companies will have implemented emergency measures to provide additional support to customers, such as video calls to show customers in real time how to perform instrument diagnosis or repair tasks that previously only engineers did. Although these show excellent flexibility and initiative and provide essential help, it is important that regulatory requirements related to training records be considered, or the work performed may be hard to defend during the next audit.
The problem, resolution, and requalification work must also be recorded in the instrument log book in chronological order as required by 21 CFR 211.182 [4], EU GMP Chapter 4.31 (20) and discussed in a “Focus on Quality” column in Spectroscopy (21).
Online Supplier Help
Don’t forget that there are suppliers who provide on-line technical support and forums where remote assistance and discussions or troubleshooting guides to diagnose problems, some of which are shown below:
Update Your Qualification Procedures
Grace periods can only be extended for so long, as mentioned earlier in this article. Therefore, your procedures will need to be updated and justified to perform any extension without a PM service and requalification of your chromatographs. Whatever is done, remember that under 21 CFR 211.160(b) (4)
and ICH Q7/EU GMP Part 2
clause 11.12 (22), any work must be scientifically sound.
A Late Addendum: MHRA Advice for Calibration and Service Support During COVID-19
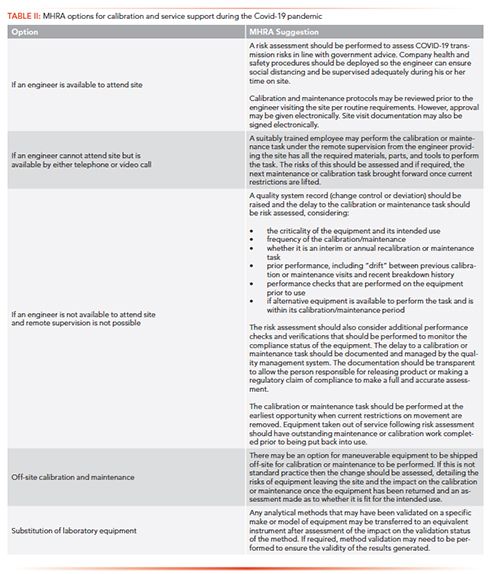
After this article was submitted for publication, the MHRA published advice on its website about what to do if it is not be possible for service personnel or external engineering support to operate as normal. A number of options are available to manufacturers and GxP laboratories (23), and these options are reproduced in Table II. Most of the options, except shipping an instrument to the supplier or service agent, were discussed above. With a module swap, it is essential that records and documentation are updated and, if applicable, a change control initiated. When the chromatograph has been shipped back to the supplier for service and qualification, the laboratory needs to test the instrument with appropriate qualification tests, such as IQ and sufficient OQ, to confirm that the chromatograph is sited correctly and has not been handled poorly in transit.

Returning to Normal?
What happens when we return to normality? If a laboratory has deviated from approved SOPs that are designed to ensure the safety and efficacy of drugs, without justification, there will be a regulatory reckoning. Instead of blaming COVID-19 for a lack of compliance, companies who have a well thought out policy of appropriate risk-based changes that justify controlled deviation from procedures will fare much better.Inspectors expect to see compliance maintained (9) even in these difficult times. Therefore, relying on effective risk management and sound science is the only way to achieve this, especially if service engineers are conspicuous by their absence.
Irrespective of the risk control strategy (acceptance versus mitigation), a laboratory should commit to reassessing all data and analytical results generated on each instrument between the date of an OQ lapsing until the date when it was maintained and requalified. In addition, you might want to consider repeating some tests for critical products to confirm that results generated using an out-of-qualification chromatograph are similar to those obtained after requalification.
Summary
This is an unusual article for unusual times. I have spent most of my career writing about what needs to be done to comply with regulations; I’m now writing about what to do if you can’t fully comply. If an engineer cannot get to your site to service and qualify your chromatograph, the approaches described in this article provide a practical, risk-based best effort to comply until life returns to normal. Nothing in this article should be used to justify doing nothing to avoid complying until the pandemic is over. It is essential to ensure that data integrity is maintained so that quality medicines can still be produced during these difficult times.
Acknowledgments
I want to thank two people who have made suggestions and reviewed this article but wish to remain anonymous. You know who you are- thank you!
References
- R.D. McDowall, LCGC N. Am.37(1), 44–51 (2019).
- R.D. McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, Cambridge, United Kingdom, 2019).
- USP 41 General Chapter <1058> Analytical Instrument Qualification (United States Pharmacopoeia Convention, Rockville, Maryland, 2018).
- 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration, Sliver Spring, Maryland, 2008).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 3 Premise and Equipment (European Commission, Brussels, Belgium, 2014).
- Inspection Observations. 2020; Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations.
- Coronavirus Disease 2019 (COVID-19) Update: Foreign Inspections. 2020; Available from: https://www.fda.gov/news-events/press-announcements/coronavirus-disease-2019-covid-19-update-foreign-inspections
- Coronavirus (COVID-19) Update: FDA Focuses on Safety of Regulated Products While Scaling Back Domestic Inspections. 2020; Available from: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-focuses-safety-regulated-products-while-scaling-back-domestic.
- New arrangements for MHRA Good Practice (GxP) inspections due to coronavirus (COVID-19) 2020; Available from: https://www.gov.uk/government/news/new-arrangements-for-mhra-good-practice-gxp-inspections-due-to-coronavirus-covid-19--2.
- Guidance for Good Laboratory Practice (GLP) facilities in relation to coronavirus (COVID-19) 2020; Available from: https://www.gov.uk/guidance/guidance-for-good-laboratory-practice-glp-facilities-in-relation-to-coronavirus-covid-19.
- D. Churchward. MHRA Inspectorate Blog: MHRA Good Practice (GxP) inspections during the COVID19 outbreak. 2020; Available from: https://mhrainspectorate.blog.gov.uk/2020/03/23/mhra-good-practice-gxp-inspections-during-the-covid19-outbreak/.
- FDA Guidance for Industry - Notifying FDA of a Permanent Discontinuance or Interruption in Manufacturing Under Section 506C of the FD&C Act (Food and Drug Administration, Sliver Spring, Maryland, 2020).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 6 Quality Control (European Commission, Brussels, Belgium, 2014).
- P. Baker, Personal Communication. 2019.
- P.A. Smith and R.D. McDowall, LCGC Europe 28(2), 110–117 (2015).
- P.A. Smith and R.D. McDowall, Spectroscopy35(4), 15–23, 50 (2020).
- P.A. Smith and R.D. McDowall, LCGC Europe 32(1), 28–32 (2019).
- R.D. McDowall, LCGC N. Am.37(5), 312–316 (2019).
- R.D. McDowall, LCGC N. Am.38(4), 233–240 (2020).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation (European Commission, Brussels, Belgium, 2011).
- R.D. McDowall, Spectroscopy 32(12), 8–12 (2017).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) guidelines, Part 2 - Basic Requirements for Active Substances used as Starting Materials (European Commission, Brussels, Belgium, 2014).
- Guidance for Manufacturers and Good Practice (GxP) Laboratories on Exceptional Flexibilities for Maintenance and Calibration during the Coronavirus COVID-19 Outbreak, Available from: https://www.gov.uk/guidance/guidance-for-manufacturers-and-good-practice-gxp-laboratories-on-exceptional-flexibilities-for-maintenance-and-calibration-during-the-coronavirus-co?utm_source=369ad22e-bfe9-426b-8b98-088e0d3c0e31&utm_medium=email&utm_campaign=govuk-notifications&utm_content=weekly
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com








; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")




. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

