Column Pressure Considerations in Analytical HPLC
LCGC Europe


In this instalment of "Column Watch", columnist Ron Majors examines the role of pressure in high performance liquid chromatography (HPLC) from two viewpoints: the impact of the ultrahigh pressures encountered in ultrahigh-pressure liquid chromatography (UHPLC) on chromatographic parameters and increases in column pressure encountered in normal daily use. The latter is of more practical consequence to HPLC users because increased back pressure usually implies that something has gone wrong with the column. Pressure increases as a result of physical and chemical contamination are explored and practical approaches to solve these problems are suggested.
Ronald E. Majors, Agilent Technologies, Wilmington, Delaware, USA.
When considering the resolution equation in high performance liquid chromatography (HPLC), we think of efficiency, selectivity and retention factors but there is no obvious, direct mention of pressure. Yet pressure is one of the most important parameters in successful HPLC. The role of pressure is to drive the mobile phase through the small particles packed into the chromatographic column. To do this, we must employ a high-pressure pump that can provide sufficient hydraulic pressure to overcome the resistance of the packed bed. Until recently, the upper pressure limit was not a major discussion point because pumps that provided up to 400 bar were freely available. Only when the column, which normally operated well below the upper pressure limit of the pump, started to give an abnormally high back pressure near this upper limit, did chromatographers get concerned about pressure. In this instalment of "Column Watch", I will cover two aspects of dealing with the pressure experienced by HPLC columns. One consideration will be a discussion of the pressure generated by going to smaller particles, a current trend in analytical HPLC. The second consideration, and perhaps more important to the average user, will be pressure changes dictated by changes in the column itself. It is the latter issue that concerns many chromatographers because increased column back pressure usually implies that something has gone wrong with the column. In some instances, the column can be dead and will have to be replaced. In other instances, the column might be salvageable. I will look at some of these issues and provide some solutions to handle this type of back pressure problem.
Column Back Pressure Equation
Just to make sure that there is an understanding of pressure terms, Table 1 provides some of the terms that are used in HPLC. The most commonly accepted unit of pressure in recent years has been bar, but psi, atm and kPa are thrown around. These interchangeable terms sometimes have caused confusion.


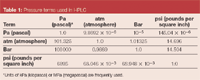
Table 1: Pressure terms used in HPLC
Now, let us look at the equation (Equation 1) that governs back pressure in a packed bed.

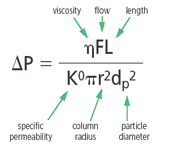
Pressure is directly proportional to viscosity (η), column length (L), and flow rate (F). In practice, the viscosity of a fluid usually manifests itself as a resistance to flow. So if a high-viscosity liquid is used as the HPLC mobile phase, a higher pressure will be observed. Thus, when a chromatographer is choosing a mobile phase component, the lowest viscosity mobile phase should be chosen. For example, in reversed-phase HPLC, if one has a choice for the organic portion of the mobile phase, then acetonitrile would be chosen over isopropanol because of its lower viscosity. Of course, there are other considerations in choosing the organic solvent such as solvent strength, polarity, analyte solubility, analyte compatibility and so on.
Column length is an obvious factor in back pressure. A longer column will require considerably more force to move a liquid through the packed bed. Sometimes, chromatographers must resort to increasing the column length to achieve a better separation. If the column becomes too long, then the pressure might exceed the capability of the pump and other adjustments to the operating conditions must be made. For example, the third component of the numerator in Equation 1 is F. Often, when the chromatographer desires to speed up the separation, increasing the flow-rate is a simple parameter to adjust. However, too high of a flow-rate can result in overpressuring the pump, so there is a compromise. If the pressure becomes too high, then reducing the flow-rate is an alternative to keep the operating pressure below the upper limit of the pump. As in all aspects of chromatography, there must be a balance of these operating parameters.
In Equation 1, the biggest factor that influences pressure is the packing particle diameter dp. The particle diameter has a big impact because pressure is inversely proportional to dp2. Decreasing the particle size by half increases the pressure by a factor of 4. Thus, if one were operating a column at a pressure of 125 bar, and if all other chromatographic parameters stayed the same, the pressure would increase to 500 bar, which exceeds the operating pressure of all but the newest generation of HPLC pumps. Fortunately, when the particle diameter of the packing inside the column is decreased, one can shorten L without sacrificing much resolution. By shortening the column length, the pressure does not increase as much. Recently, there has been a movement to decrease the particle diameter of commercial columns to below 2 μm and pump pressures of older generation pumps might be limited, especially for columns 100–150 mm in length.
Higher column back pressures have been encountered because of the increased use of sub-2 μm particles, resulting in back pressures that are higher than previously experienced. Thus, the use of temperature to lower solvent viscosity has become fashionable. With an increase in temperature, column back pressure decreases because of a decrease in η. In addition, efficiency increases as a result of more rapid solute mass transfer and retention decreases as solutes favour the mobile phase. Selectivity (relative retention) can also be affected with changes in temperature.
As long as the column maintains a steady back pressure, there is generally no need for concern. Of course, when a gradient is performed, it is common for the back pressure to change because of viscosity changes of the mobile phase as solvents are mixed during gradient elution. When the column begins to experience significantly increasing back pressure, that is a bad sign. We will explore this aspect further later in this article.
The Effect of High Pressure on Chromatographic Parameters
With the increase in chromatographic pressure, a closer look has been taken at how these pressures will impact the chromatographic experiment. Although there have been many studies over the years using high pressures in liquid chromatography,1–15 commercial instrumentation has only recently been made available that can make use of small particles and their greater efficiency gains. Pumping systems are now available commercially that can achieve up to 20000 psi (1380 bar). Commercial columns packed with particles down to 1.5 μm are also available. Some of these sub-2 μm packed columns are available in lengths up to 150 mm. To illustrate the impact of these smaller packings when packed into longer columns, Table 2, taken from reference 14, compares the expected performance and pressure requirements of 25 cm-long columns, packed with particles ranging from 0.75 to 5 μm in size, operated at the optimum flow velocity for a small organic analyte. Reducing the particle diameter from 5 μm to 1 μm yields a five-fold increase in theoretical plates and a five-fold decrease in analysis time. However, the pressure requirements increase 125-fold. To take full advantage of the performance of small particles, these pressure limitations must be overcome.


Table 2: Pressure requirements and performance expected for differing stationary phase particle diameters in a 25-cm long column. Values calculated for an analyte with k = 2, DM = 6.0 x 102â6 cm2/s, and a mobile phase viscosity (η) = 1.0 cP. (taken from reference 14)
The most intensive studies on high-pressure HPLC have been performed in the laboratories of Jorgensen,3,5,7,8,14 Lee4,6,13,15 and Colon.11 Jorgensen used the term ultrahigh-pressure LC (UHPLC) to describe pressures in excess of the commonly used pressures at the time of the publication. This term seems to have stuck for work being performed with sub-2 μm packed columns.
In 2005, Martin and Guiochon published a more rigorous study that addressed some of the issues raised by Jorgensen in his UHPLC papers,16 such as a change in the retention factor with pressure increases. In commonly used chromatographic conditions of several hundred bar, many of the normal chromatographic parameters are affected very little. However, when the conditions wander into the 1000+ bar range, high pressure induces changes in the physical-chemical properties of solvents, solutes and column packings. For solutes, molar volume, solubility and diffusion coefficients change. For solvents, the melting point increases, and density and viscosity increase. For the column, geometric deformation of the bed and column tube occurs. As pointed out earlier,14 high pressure gives rise to significant viscous heating inside the column, which can give rise to changes in radial and axial velocity profiles that can result in band broadening, the temperature change can shift k values and local changes in viscosity, density and other parameters can occur. The generated heat (power), equal to the product of the flow-rate times the pressure, can cause significant temperature gains that increase with ΔP2 for a thermostated column and even more strongly in an insulated or poorly temperature-controlled environment.17 Jorgensen showed several watts of power dissipation can occur in a conventional HPLC column,3 necessitating the use of smaller internal diameter columns that dissipate the heat more effectively in UHPLC. The reader is referred to the previous references to gain a more thorough understanding of the effects of ultrahigh pressure on the chromatography.
What Causes Column Pressure Changes?
Mechanical causes of pressure changes: Basically, to diagnose pressure problems, it pays to take a look at the basic construction on an HPLC column. Figure 1 shows a typical HPLC stainless steel column configuration. A typical analytical column has an outer diameter compatible with compression fittings (1/4 in.) and an inner diameter of 4.6 mm. The column usually consists of a stainless steel tube (although other materials such as PEEK or glass are used). The packing is contained in this tube and is held in place by endfittings at either end. The endfittings used in HPLC columns are most frequently compression fittings containing a ferrule and a frit. The endfittings must withstand the packing and operating pressure of the column.

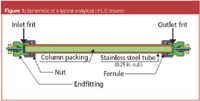
Figure 1
An important element in the endfitting is the frit. One has an inlet frit at the top and an outlet frit at the bottom. Sometimes the frit is pressed into the column rather than the endfitting. The frit has a diameter that must match the column tubing outside diameter. It must be thick enough to have good mechanical strength, yet thin enough to ensure mobile phase and sample permeability. The porosity of the frit is how much "empty space" is in the frit. It must not have too much porosity so that it is not mechanically stable or so little porosity that it becomes a restriction. Finally, just like the packing material being held, there is a pore diameter that is the average diameter of the openings in the frit surface.
The job of the outlet frit is to contain the packing material. During the slurry packing process for an HPLC packing material, this frit and the column packing suspended in the slurry are subject to very high pressure. The porosity and mechanical strength of the outlet frit are critical to contain the on-rush of slurry during the packing process. Packing pressure higher than the normal operating pressure is essential to form a tight bed. Packing particles must withstand the high packing velocity that occurs during the packing process. If the particles are not mechanically strong, they can be fractured, resulting in fines and potential plugging problems down the line. If particle breakage occurs, the column will show an abnormally high back pressure after it is packed when it is tested. Most manufacturers have pressure specifications and will reject a column that displays abnormally high pressure after packing. However, there can be instances in which particles are fractionated within the packed bed during use. An example of the latter situation might be when using a high pH mobile phase on a silica gel-based column. High-pH buffers can cause dissolution of the packing and fines can be created. The fines might eventually work their way down the packed bed and lodge in the outlet frit causing plugging to occur. Remedying this situation presents a difficulty because removing the bottom fitting can disturb the packed bed and ruin the column.
Because column-packing particles contain a distribution rather than discrete sizes, the porosity of the frit must be sufficient to retain the lower end of the particle size distribution. Otherwise, some of the packing particles could pass through the frit. As mentioned earlier, the outlet frit itself has its own pore size distribution because the frit is constructed of sintered stainless steel particles. The pores in the frit structure are the spaces between these sintered particles and they allow liquid to pass freely but prevent packing particles from doing so. For a 5 μm particle, a typical particle size distribution of this packing might range from 3 μm to 7 μm, with 5 μm being the average. Thus, the frit's average porosity must be smaller than the smallest particle size in the packing particle size distribution. So for the previous example, one would choose a 2 μm frit to use for the outlet, below the 3 μm particle size, the low end of the distribution. As the particle size decreases, the frit porosity must be reduced proportionally. Smaller porosity frits will retain smaller packing particles but can also be plugged easier or will show higher back pressure themselves.
The inlet frit also serves to contain the packing material but not to the extent of the outlet frit. The inlet frit is not subjected to the on-rush of the packing slurry because it is affixed after the column has been packed at high pressure. However, during operation, the inlet frit is exposed to incoming mobile phase and incoming liquid sample from the injector. If these incoming liquids are not entirely clean, especially with regard to dust and other particulates (for example, seal materials, stainless steel filings, buffer salts, solvent impurities and so forth), the inlet frit is the first porous bed that is encountered and it becomes an excellent filter that prevents the particulates (larger than the pores) from entering the column and perhaps plugging the packed bed. However, the frit itself can become plugged and one sees a rise on the pressure readout.
Usually, plugging of the inlet frit is not a catastrophic occurrence — it happens gradually until the pressure rises sufficiently to shut down the pump. Particulates build up on the inlet frit and actually can lodge within the porous matrix, preventing the free flow of mobile phase. In some instances, if salts are precipitated in the column inlet, the head pressure can rise dramatically rather quickly. Sometimes particulates entering the inlet from the tubing connecting the injector and the column build-up in the centre of the inlet frit and do not immediately cause a build-up of pressure. However, this build-up can only partially block the inlet frit causing a nonuniform distribution of sample and peak asymmetry.
What are Sources of Particulates?
There are a number of sources of particulates (solids) that can plug the inlet frit. Particulates can be present in the samples being injected. During sample preparation, there are often several sample preparation techniques employed such as extraction, evaporation, precipitation and so on. Particulates can come from dust, salt residues, filter paper and other sources that contaminate the sample. Particulates can result from the normal wear of the injection valve rotor seal, which consists of polymeric materials such as polyimide (Vespel, DuPont, Wilmington, Delaware, USA), ethylene–tetrafluoroethylene copolymer (Tefzel, DuPont) and PEEK. With time, these rotor seals can give off very small pieces of polymeric material that can lodge on the inlet frit. Particles in the mobile phase can also be culprits. HPLC-grade solvents purchased from reliable manufacturers are usually prefiltered at the factory and do not need further filtration. However, aqueous and organic mobile phase mixtures that one prepares in the laboratory such as buffers and solvent mixtures are subject to the introduction of particulates during the weighing, pipetting, volumetic transfer and salt dissolution processes.
Pump seals and check valves are another source of particulates. These consumable items are subject to day-in and day-out mechanical stress and eventually wear out. The devices contain polymeric sealing materials that are subject to the same wear problems as injector rotor seals. The particulates can be small enough so as to pass through some of the HPLC system components (for example, mixers, pulse dampers and tubing) but large enough to lodge in a porous frit at the inlet to the column.
An overlooked source of particulates is salt precipitates. Aqueous buffers are often used in reversed-phase, ion-exchange and aqueous size-exclusion chromatography. Many buffer salts are insoluble in mobile phases with high organic content or 100% organic. If the aqueous–organic mobile phases are created in the gradient pumping system, at high organic levels there might be the possibility of precipitation or emulsion formation upon mixing. It is best to externally check the highest organic solvent composition added to the aqueous solution to make sure that no cloudiness occurs (indicative of salt precipitation). Such a check for precipitants can also prevent seal wear within the flow system. Also, do not forget that the sample solvent can be a source of salt precipitation. If the sample is loaded with salt and it is injected into a high-organic mobile phase, the salt in the sample can precipitate. In this instance, a simple desalting procedure can be used to remove this salt. Vice versa, if the mobile phase is high in salt and a large volume of organic solvent is used for the sample injection, salt in the mobile phase can come out of solution.
How to Cope with Particulates and Plugged Inlet Frits
The best way to deal with particulates is to remove them before they reach the inlet frit. Samples and even standards should be filtered using HPLC membrane syringe filters, available from a variety of manufacturers. Clean liquid samples prevent the blocking of capillaries, frits and the column inlet. In addition, clean samples result in less wear and tear on the critical moving parts of injection valves. Sample filters come in a number of different porosities and diameters. For use with 3.0–3.5 μm and 5.0 μm columns, a 0.45 μm porosity filter is suggested. For the newer sub-2 μm columns, a 0.20 μm porosity filter would be best because frits used on these columns are generally much smaller than those used on conventional 5 μm columns. If sample volume is limited, use the smallest possible filter diameter such as 13 or 25 mm.
An important consideration is the choice of membrane. Choice of membrane is dictated by injection-solvent and analyte compatibility with the polymeric material. Syringe filters made from materials such as regenerated cellulose, PTFE, nylon, cellulose nitrate and cellulose acetate are common in the HPLC laboratory. Readers are directed to syringe-filter manufacturers' literature that often have solvent compatibility tables or to an excellent article in a previous issue of LCGC N. Am.18
For mobile phases, especially self-made buffers, filtration is also recommended. Clean mobile phases reduce wear on instrument parts (for example, check valves, piston seals and autosamplers). Again, make sure that the filter membrane used is compatible with the mobile phase solvent being filtered. Simple vacuum filtration flask systems are available commercially. An added benefit is that in vacuum filtration, the mobile phase is degassed also but most modern HPLC systems have in-line degassing as a standard feature. Mobile phase filters can also be placed in the solvent reservoir. It is attached to the end of the reservoir tube between it and the pump inlet. Mobile phase reservoir filters generally have rather large porosities (greater than 2 μm) because they cannot provide a restriction to the pump inlet. Otherwise, cavitation can occur.
A popular approach to ensure clean samples or mobile phases is to install an in-line filter (see Figure 2). These days, in-line filters have minimal dead volume, so they provide minimal contribution to band broadening. The filter can be inserted between the injector and the analytical column (column prefilter) or between the pump and the injector. When placed between the injector and the column, the filter protects the column from mobile phase as well as sample particulates. When placed between the pump and the injector, the filter protects against mobile phase particulates only. Some in-line filters have replaceable filter elements and are finger-tightened. For samples that are sensitive to stainless steel, metal-free in-line filters are available. In-line filters must be able to hold up to the column operating pressure because they are installed on the high-pressure side of the pump.

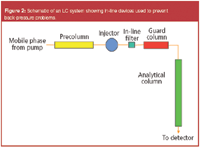
Figure 2
Another particulate removal technique is the use of a guard column (see Figure 2). Normally, a guard column is recommended to prevent chemical not mechanical contamination of the analytical column (see the following). It is placed between the injector and the HPLC column. A guard column is a shorter version of the analytical column and should normally be packed with the same packing material. Because the guard column is frequently constructed in a similar manner to the analytical column, it is the first system component that particulates might meet (if no in-line filter is installed). Thus, it would become plugged before the analytical column is plugged. Because the cost of the guard column is a fraction of the analytical column, it is generally discarded and replaced when its pressure rises or it becomes contaminated.
Once the inlet filter becomes blocked and the column pressure rises unacceptably, there are a couple of approaches that can be tried to rectify the situation. The most popular approach is to backflush the column by removing it from the chromatograph and attaching the inlet line from the injector to the column exit. The idea here is to dislodge accumulated particulates by flushing them out of the inlet frit. Do not connect the column to the detector during backflushing to prevent accumulated particles from being flushed into the detector flow cell and perhaps causing problems there. The column outlet (formerly the inlet!) should be directed to waste. The initial flow-rate should be lower than the normally used flow-rate but can be increased as the particulates are removed. Sometimes, this procedure does not do the trick because the particulates have become embedded within the porous elements of the frit.
One word of caution: make sure that the inlet frit's porosity is lower than the particle size distribution of the packing in the column (as discussed earlier). Some manufacturers use a higher porosity frit (2 or 5 μm porosity) at the inlet to the column to prevent early plugging. If such a column is backflushed, there is a chance of also flushing out some of the packing particles during the process. Check the column instruction or data sheet or call the manufacturer to make sure that their columns can be backflushed. If the column has an arrow on its side, this sign might be indicative of a column not to be backflushed. In general, any well-packed column should be operable of flow in either direction.
A second approach to solving a plugged inlet frit is to replace the endfitting. In general, I would not recommend doing this because the packing material that is in contact with the frit will almost always be disrupted because it sticks to the frit (inlet fitting) that is removed. Replacing this disrupted (removed) packing material with some additional packing will not provide a homogeneous bed, and column efficiency will be impaired. Instead, if the backflushing cannot be accomplished or is not successful, it is probably best to discard the column.
One other possibility of an increase in column pressure is a blocked flow line in the system between the pump outlet and the head of the column. A blocked line could result from a particulate lodging in the tiny internal diameter tubing that is now being used to decrease system dead volume required for the newer, high-efficiency columns. A systematic removal of system components from the column backwards through the connecting line from the injector, any in-line filters or guard columns, the injector itself and prior system components should eventually find the blocked element. Obviously, if the analytical column is removed from the system and the pressure readout is still high, the column is not the culprit so work backwards accordingly. Be careful when performing this systematic removal at high pressure just in case a blockage is quickly dislodged because solvent can be sprayed from the component.
Chemical Causes of Pressure Build-up
So far, I have covered mechanical reasons (for example, particulates or blockages) why column pressure can increase. An equally strong possibility is pressure build-up caused by chemical contamination of the packed bed. Just like mechanical contaminants, chemical contaminants can arise from a variety of sources — samples, sample matrices, mobile phases or system components. The most probable source of chemical contamination is from the sample, especially if insufficient or no sample preparation has been used to treat a complex sample.
Usually there are compounds in the sample matrix that are of no interest to the analyst but are present nevertheless. High molecular weight compounds, salts, lipids, waxes and fatty compounds, humic acids, hydrophobic proteins and other biological compounds are but a few of the myriad possible substances that can come in contact with an HPLC column during its use. These materials can have lesser or greater retention than the analytes of interest. Those that have lesser retention such as salts will usually be eluted from the column at the void volume. These undesired interferences might or might not be observed with the detector being used and, if detected, show up as chromatographic peaks, blobs, baseline upsets, sometimes even as negative peaks. If sample matrix components are strongly retained on the column and if the mobile phase solvent composition itself never becomes strong enough to elute them, over the course of many injections, these adsorbed or absorbed compounds will accumulate, usually at the head of the column. Such behaviour is observed more often when isocratic conditions are used. Sample compounds that are of intermediate retention can be eluted slowly and show up as wide peaks, baseline disturbances, or as baseline drift. With gradient conditions, because the column is subjected to a stronger mobile phase during the run, sometimes these contaminants are removed in the latter stages of the gradient run.
On occasion, the sorbed sample (or mobile phase) components build up to such high levels they can begin to act like a new stationary phase. Analytes can interact with these impurities that now contribute to the separation mechanism. Retention times might shift and tailing can occur. If sufficient contamination occurs, the column back pressure can build up to intolerably high levels, overpressuring the pump and perhaps causing the column to settle and void, depending upon where the blockage occurs.
It is this latter instance that I will explore in this section. Noneluted sample components usually build up on the HPLC inlet column packing and, if unchecked, can cause these pressure build-ups. The best way to remove chemical contaminants is to flush the column with a solvent that will dissolve the contaminants, yet will not harm the column packing itself. For example, precipitated proteins are often removed from a polymeric column by flushing the column with strong base, perhaps of pH 13 or 14. However, a silica gel-based column would most probably be harmed in this process, unless the column is a special reversed-phase column designed for high pH.
The best way to clean a column is to backflush (not forward flush) the column with a solvent or series of solvents that will remove the contaminants causing the pressure build-up. Note that forward flushing is not recommended because sparingly soluble substances can take a long time to flush the entire length of the column and can become spread out during the process. Instead, in the reversed configuration, the path length from the inlet "chemical plug" to the column exit is much shorter and the washing time greatly reduced.
Several years ago, I wrote an article on the cleaning and regeneration of reversed-phase HPLC columns.19 In this article, I discussed the approach for washing silica- and polymeric-based reversed-phase columns to remove protein and other residues. In addition, special techniques for cleaning reversed-phase columns when simple solvent washing does not work were covered. Rather than repeating the contents of the article here, I refer the reader to the original publication. Just remember, before washing any column used with a salt buffer, first remove the salt by flushing with a salt-free mobile phase of equivalent composition. A similar article on the regeneration of biopolymer columns was published many years ago but is still applicable today.20
For normal-phase columns such as silica gel, cyano, or diol, a similar procedure can be used in which progressively stronger solvents are used to wash contaminants from the column. For these columns, we have found that washing the column with 20 column volumes (a 250 mm × 4.6 mm column has a column volume of about 2.5 mL) of a 50:50 mixture of hexane–chloroform followed by methanol, methylene chloride, or 100% ethyl acetate seems to do a good job. Isopropanol can also be used; it seems to have good solubility properties for fatty compounds. Be careful not to run too high of a flow-rate during the washing experiments, as isopropanol's viscosity is quite high.
Microbial growth often occurs in buffers and aqueous mobile phases, especially if they are permitted to stand around for long periods of time at room temperature. Particulates or the bacteria from microbial growth can plug the column inlet or lodge on the column packing itself.
Another overlooked source of chemical contamination is the use of the wrong rotor material in the injection valve for noncompatible solvents or pH. For example, Vespel, a frequently used polymeric material in rotary injector valves, is not compatible with high-pH buffers. Prolonged use of such a valve under basic conditions will result in increased wear and earlier failure. Instead, in this situation, a Tefzel valve core material is more compatible. Consult with the valve or instrument manufacturer on solvent compatibility of instrument parts.
Prevention of Chemical Build-up on HPLC Columns
The best way to prevent chemical contamination is to remove these undesired sample components before they reach the analytical column. Proper sample preparation procedures to remove matrix and interfering compounds are paramount to extending column life and in preventing pressure build-up. There are many procedures used for sample clean-up.21,22 For liquid chromatographers, solid-phase extraction (SPE) and liquid–liquid extraction are among the more popular. Through the removal of undesirable compounds, method development is also much easier, column overloading can be avoided and chromatographic resolution can be increased.
The use of a guard column (Figure 2) is recommended as a safeguard to prevent chemical contamination of the analytical column. The guard column should contain packing that is the same as that used in the analytical column. Otherwise, it can affect the chromatographic separation. If, for some reason, a guard column is to be used that contains a different bonded phase, then the bonded phase should be a weaker (not a stronger) sorbent than the analytical phase it is to protect. The purpose of the guard column for this instance is to adsorb (or absorb) those strongly retained components from the sample that will foul the stationary phase on the analytical column. As the contaminants build up and the pressure on the guard column rises, it is replaced. Because the guard column is much lower cost than the analytical column that it protects, it is probably not worth the time to clean this column to reclaim it, although there is no reason why this could not be done. The guard column can serve to chemical contaminants introduced by the mobile phase, although normally mobile phase components (for example, buffer salts, aqueous and organic solvents, acids, bases, additives and so on) are selected in the purest available state.
One other device, shown in Figure 2, that has been used for column protection is the precolumn. This column is sometimes used to presaturate the mobile phase with dissolved silica when the mobile phase pH exceeds that recommended for the bonded silica column. Many silica-based HPLC columns have an upper pH limit of 8–9 because of dissolution of the underlying silica by the hydroxide ion. The idea of the saturator column, packed with coarse silica particles, is to condition the incoming mobile phase with dissolved silicate, retarding silica dissolution of the analytical column. Usually, a 0.2 μm in-line filter is placed just after the precolumn to exclude any dissolved particles from the injector and the column inlet. However, the added volume of a precolumn adds greatly to the gradient delay volume, adds back pressure to the system, and because of this additional pressure the analytical column pressure cannot be determined alone. For those reasons, the use of precolumns is not popular anymore. Rather, more chemically stable analytical columns should be used instead. However, sometimes precolumns are used for isocratic analysis where the additional volume is of minor consequence.
To prevent microbial growth, it is recommended that pure aqueous buffer be discarded at the end of the day. When preparing buffer, use only the amount required for one day's use and place the remainder in the refrigerator. To retard microbial growth, some recommend adding 200 ppm of sodium azide to the aqueous mobile phase. However, azides are very toxic and potentially explosive. Alternatively, if one is not using 100% aqueous at the beginning of the gradient, then the addition of 10% organic to the aqueous solvent (solvent A) is sufficient to retard bacterial growth.
Column Pressure Decreases or Fluctuations
Up to this point, I have discussed the normal situations in which the column pressure increases. Column pressure can also decrease or fluctuate, but these pressure changes are not associated with the packed bed or the frits. Column pressure decreases are the result of hardware problems such as pump cavitation, leaking pump check valves or piston seals, leaking injector seals or leaks somewhere in the system (for example, fittings). Usually leaks are rather obvious because liquid will be spotted around the leaking component. Pressure fluctuations are most probably a result of pump cavitation or a bubble in the pump.
Conclusions
If this instalment of "Column Watch" I have investigated pressure from two aspects: the impact of ultrahigh pressure on physiochemical parameters and pressure changes as a result of mechanical or chemical blockages. The former aspect was to make users aware of how pressure can impact chromatographic performance because of the recent trends of increased use of ultrahigh pressures. The latter aspect of pressure changes is more immediate for most users because it affects column life and back pressure of commonly used analytical columns. Pressure blockages from particulates can be prevented by the installation of in-line devices such as filters and guard columns or by sample and mobile phase filtration. Pressure increases as a result of chemical contaminates in the packing can be handled by thorough washing of the columns, use of proper sample preparation techniques and by the use of disposable guard columns.
"Column Watch" editor Ronald E. Majors is business development manager, Consumables and Accessories Business Unit, Agilent Technologies, Wilmington, Delaware, USA and is a member of the Editorial Advisory Board of LCGC Europe.
Direct correspondence about this column to LCGC Europe, Advanstar House, Park West, Sealand Road, Chester CH1 4RN, UK, E-mail: amatheson@advanstar.com
References
1. B.A. Bidlingmeyer et al., Sep. Sci., 4, 439–446 (1969).
2. B.A. Bidlingmeyer and L.B. Rogers, Sep. Sci., 7, 131–157 (1972).
3. J.E. MacNair, K.C. Lewis and J.W. Jorgenson, Anal. Chem., 69, 983–989 (1997).
4. J.A. Lippert et al., J. Microcolumn Sep., 11, 631–643 (1997).
5. J.E. MacNair, K.D. Patel and J.W. Jorgenson, Anal. Chem., 71, 700–708 (1999).
6. N. Wu et al., J. Microcolumn Sep., 12, 462–469 (2000).
(7) J.W. Jorgenson et al., "Gradient Elution Separation In Ultra-High Pressure Liquid Chromatography," L–0901, presented at HPLC 2000, Seattle, Washington, USA, 24–30 June (2000).
8. L.T. Tolley, J.W. Jorgenson and M.A. Moseley, Anal. Chem., 73, 2985–2991 (2001).
9. N. Wu, J.A. Lippert and M.L. Lee, J. Chromatogr. A, 911, 1–12 (2001).
10. Y. Shen et al., Anal. Chem., 73, 3011–3021 (2001).
11. J.M. Cintrón and L.A. Colón, Analyst, 127, 701–704 (2002).
12. D.C. Collins, Y. Xiang and M.L. Lee, Chromatographia, 55, 123–128 (2002).
13. Y. Xiang et al., Chromatographia, 55, 399–403 (2002).
14. A.D. Jerkovich, J. Scott Mellors and J.W. Jorgensen, "Recent Developments in LC Column Technology", R. Majors, Ed., LCGC Eur., 16(6a), 20–23 (2003).
15. Y. Xiang et al., J. Chromatogr. A, 983, 83–89 (2003).
16. M. Martin and G. Guiochon, J. Chromatogr. A, 1090, 16 (2005).
17. K. Broeckhoven et al., Lecture L22.01, HPLC 2007, Ghent, Belgium, 21 June (2007).
18. R. Lombardi, "Current Trends and Developments in Sample Preparation", R. Majors, Ed., LCGC N. Am. Special Supplement, May, S47–S52 (1998).
19. R.E. Majors, LCGC N. Am., 21(1), 19–26 (2003).
20. C.T. Wehr and R.E. Majors, LCGC Int., 1(4), 10–17 (1988).
21. R.E. Majors and D. Hardy, LCGC Eur., 5(6), 10–16 (1992).
22. R.E. Majors, LCGC Eur., 16(2), 71–81 (2003).

















Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.

Characterizing Polyamides Using Reversed-Phase Liquid Chromatography
May 5th 2025Polyamides can be difficult to characterize, despite their use in various aspects of everyday life. Vrije Universiteit Amsterdam researchers hoped to address this using a reversed-phase liquid chromatography (RPLC)-based approach.


New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

