Chromatography Forum — Use the Braintrust
LCGC North America
John Dolan explores how an on-line discussion group can be a great source for free consultation from experts in the field.
Chromatography Forum is an on-line discussion group sponsored by LCGC, LC Resources (Walnut Creek, California), and some other vendors. There are forums for liquid chromatography (LC), gas chromatography, hyphenated techniques, data systems, and student projects. As of the time I'm writing this column, there are more than 1600 topics and 8000 posts in the LC section, so you can see that this is an active discussion group. The Forum is administered by Tom Jupille, of Separation Science Associates, who helps to keep the discussion constructive and not a vendor-bashing gripe session. One thing that impresses me is the number of experts who regularly participate in the Forum. What a wonderful way to get free consultation from a variety of experts in a timely manner. In this month's installment of "LC Troubleshooting," I've used one thread from Chromatography Forum to illustrate the resources available to participants. (In the following sections, I've identified various participants by their initials, for example, RB, to help you keep track of the players.) To participate in the Forum, either as an observer or a contributor, log on to www.chromforum.com. It will be worth your time.
The Problem: Split Peaks
The problem that RB reported is illustrated in Figure 1 for a sample containing a secondary amine with an acidic salt counter ion as the compound of interest plus related impurities. The method comprised a gradient of 0.1% formic acid (mobile phase A) and 0.1% formic acid in methanol (mobile phase B) run from 30% B to 70% B in 8 min, followed by an isocratic hold at 70% B. I assume that the column was a C18, type B silica column, and from the chromatogram, I guess that a 250 mm X 4.6 mm was used at a flow rate of 1 mL/min. The system dwell volume was ≈1.2 mL, the column was operated at 29 °C, and the UV detector was set to 254 nm. The sample was diluted to ≈0.2 mg/mL in 60:40 water–methanol; lower concentrations of methanol resulted in poor extraction of the impurity peaks. As first identified in Figure 1, an injection volume of 40 μL gave a nicely shaped peak, whereas a 30-μL injection gave a split peak that also was broad and more strongly retained.
Injection Problems?
CPG suggested that the injection volume should be reduced to 10–15 μL. ST suggested that the injection solvent should be changed to the initial mobile phase. If the sample is injected in too large a volume of a solvent that is greater than or equal to the mobile phase strength, peak splitting and retention changes can occur. If the mobile phase is used as an injection solvent in an isocratic run, any band spreading that occurs at the top of the column will continue through the system and result in a broader peak at the detector. A good rule of thumb is to keep the injection volume no larger than 15% of the volume of the peak of interest when injecting in the mobile phase. For the well-shaped peak in the lower trace of Figure 1, the peak width is ≈0.25 min. At a flow rate of 1 mL/min, this peak width would convert to a volume of ≈250 μL; 15% of 250 μL is 40 μL, so the injection volume doesn't seem too excessive for an isocratic run (a gradient is used here). In a gradient method, if the peak migrates during the initial gradient conditions, the same problem can occur. However, if the peak is retained sufficiently by the column under the injection conditions, on-column concentration can occur so that the peak is concentrated at the head of the column and no extra broadening will be observed. The use of an injection solvent weaker than the starting gradient conditions should provide for on-column concentration.
The test of reducing the injection volume is simple and RB tried it, but no improvement was found. Injecting using the mobile phase as the sample diluent did not help, either. It also was noted that smaller injection volumes are not always practical with an impurity assay, such as this, because the peaks for the impurities will be too small to measure.
UN suggested a further experiment to test the injection volume–solvent strength question. Dilute the sample 1:1 with the initial mobile phase and double the injection volume. This would dilute the injection solvent to a concentration more like the initial mobile phase. By doubling the injection volume, the mass-on-column would remain the same, so the peaks should be the same size. RB reported that this experiment did not improve matters either. In addition, RB tried several modifications of the gradient, including placing an isocratic hold at the front of the gradient and starting at a lower %B. None of the changes solved the problem.
Other Chemistry Problems?
Several participants suggested other changes to the system chemistry that might fix the problem. NM suggested adding triethylamine to the mobile phase. This was a popular and effective solution to prevent unwanted interactions between basic compounds and the silica-based column packing of the older, type-A silica columns. These columns were quite acidic and had sufficient cation-exchange properties that basic compounds nearly always tailed. Addition of triethylamine at a concentration of ≈25 mM swamped out these active sites and gave better peak shape. However, today's newer type-B columns use a much higher purity silica that is much less acidic and has much weaker cation-exchange sites, so triethylamine rarely is necessary.
CP suggested that the mobile phase ionic strength was too low. This, also, was a problem more common with type-A columns. Higher ionic strength mobile phases tended to mask the ionic characteristics of the column.
RB did not report back on the results of any of these suggested experiments. However, my guess is that they would have little impact on the column, if it is indeed a type-B silica column.
Equipment Problems?
In one of his reports back to forum participants, RB mentioned that he tried the method on two other high-pressure-mixing systems and did not see the peak-splitting problems. This prompted JM to suggest that the problem was related to a proportioning valve malfunction, which can happen with low-pressure mixing systems. He also indicated that he had seen similar problems with bad check valves and pump seals. He suggested running a pressure recording at the same time as the gradient to see if there were pressure abnormalities associated with the runs in which peak-shape problems were observed. If a check valve or pump seal malfunctioned, a dip in the pressure should be observed.
TJ followed this suggestion, with the recommendation that a plot of the gradient output from the system should be made. This is done by replacing the A-solvent with water and the B-solvent with water spiked with 0.1% acetone; 1 m or so of 0.005-in. i.d. tubing is substituted for the column and the detector is set to 265 nm. When a blank gradient is run, the detector output should reflect the gradient shape programmed into the system. (This test also can be used to measure the dwell volume of the system.) Any abnormalities in the plot can be correlated with solvent proportioning problems related to some part of the system.
With the help of a field engineer, RB traced the problem to the inlet-line frit in the B-solvent reservoir. It was observed that bubbles occasionally were drawn into the inlet tubing. Replacement of the frit seems to have solved the problem.
Reprise
Now that the problem is solved, let's look back over it and see what we can learn, and perhaps get some ideas about solving the problem more quickly in the future. Of course, 20/20 hindsight gives us a bit of a biased view of the situation. The problem chromatograms were observed initially with smaller injections and also longer retention times. Although the smaller-injection and weaker-solvent experiments were easy to run, it might not be too surprising that these did not solve the problem. Overload, either by injection volume or injection solvent strength, should reduce the retention time because the conditions tend to wash the sample rapidly down the column before the injection solvent is diluted sufficiently for normal retention to take place. Smaller injections generally improve the conditions, but initially, 30 μL was worse than 40 μL. It turns out that the correlation of injection size with the problem was a red herring.
Later elution under the problem conditions should have pointed immediately to a solvent strength or flow rate problem. It is interesting to note in Figure 1 that the peak was eluted at ≈16 min has approximately the same time in the good and bad runs, as does the small peak at ≈12.5 min, whereas the problem peak differs by more than a minute. This suggests that whatever is happening, it is momentary in nature, because it did not affect all the peaks in the run. The recommendation to step back and perform a system check with water–acetone was an excellent one. When a problem is not solved quickly, it is a good idea to go back to basics and determine if the equipment is functioning properly or not. I recommend performing this test once every six months as part of the semiannual preventive maintenance on the HPLC system.


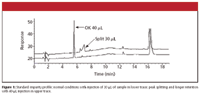
Figure 1
The problem with the inlet-line frit is not one that I've seen in this context with high-pressure-mixing systems. One needs to be careful to watch for this problem with low-pressure mixing systems, because erroneous solvent proportioning can occur if one frit becomes partially blocked. In the low-pressure mixing case, if one frit were partially blocked, it would not allow that solvent to be delivered at the desired rate, thus, changing the mobile-phase mixture. For example, consider the case in which the pump was set to deliver 50:50 water–methanol at 1 mL/min and the water frit was partially blocked. When the water-proportioning valve opened, it would deliver less than 1 mL/min — this would result in a partial vacuum being formed in the mixer. The water valve would close and the methanol one would open, and because there was no restriction in the methanol line, it would deliver more than 1 mL/min to satisfy the vacuum. The net result would be a mobile-phase mixture with more methanol than desired. With high-pressure mixing systems, the solvent mixture is controlled by the flow rates of the two pumps, so solvent always flows from both reservoirs (except at 0% B and 100% B settings). If the demand is larger than the frit allows, a bubble often forms in the tubing (called "cavitation") and results in the delivery of less of that solvent than was programmed. In my experience, the most obvious problem in such situations is the presence of bubbles in the pump, which creates noticeable pressure fluctuations. If an in-line membrane degasser were used, it might be possible that the bubbles were removed and yet the pump was still starving. Another surprise to me is that the frit problem was in the organic reservoir, not the aqueous one. Generally, the water reservoir frit fails first because the environment is more conducive to microbial growth, which can foul the inlet-line frit.
The condition of the inlet-line frit can be checked with a simple siphon test. Just disconnect the inlet tubing at the proportioning manifold (low-pressure-mixing) or the pump inlet (high-pressure-mixing) and siphon mobile phase through the line. With a typical head pressure of 0.5 m or so, I like to see the siphon deliver at least 10 times the volume that is required. For example, if you normally run the system at 1–2 mL/min, you should expect to have at least 20 mL/min through the siphon to ensure that there are no restrictions in the tubing or frits. Under these conditions, 50 mL/min from the siphon is common with a new frit. Frits are inexpensive, and it should be easy to justify replacing the frits once or twice a wear as part of a preventive maintenance program.
Free Consultation — Timely Advice
This thread in the Forum had at least a dozen participants that contributed to solving the problem. It also is interesting to note that the first posting of the problem was on Tuesday, November 29, and the problem was reported as solved on Friday, December 2. The free help of a dozen helpful experts over a period of four days to help solve your problem — what a bargain! Join the Forum and you'll learn from others. Hopefully your experience can be used to benefit your fellow chromatographers, too.
John W. Dolan "LC Troubleshooting" Editor John W. Dolan is Vice-President of LC Resources, Walnut Creek, California; and a member of LCGC's editorial advisory board. Direct correspondence about this column to "LC Troubleshooting," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail John.Dolan@LCResources.com
For an ongoing discussion of LC trouble-shooting with John Dolan and other chromatographers, visit the Chromatography Forum discussion group at http://www.chromforum.com.

















New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

