Biolayer Interferometry as an Alternative to HPLC for Measuring Product Concentration in Fermentation Broth
LCGC North America


Several key applications of biolayer interferometry in pharmaceutical development have emerged recently. Here, we evaluate its use for easuring product titer from fermentation, and compare the strengths and weaknesses of the technique to those of HPLC.
In this installment, we showcase the use of biolayer interferometry (BLI) for measuring the titer of a product: a fragment antigen-binding (Fab) fragment that has been expressed in the periplasm of E. coli. High performance liquid chromatography (HPLC)-based quantification is also performed to showcase the high-throughput characteristics of BLI versus HPLC.
The rising number of life-threatening diseases has translated into a steady increase in demand for biotherapeutics. However, biotherapeutics continue to suffer from poor affordability when compared to other pharmaceutical products. This anomaly has created a significant pressure toward increasing the productivity of biotech processes. Escherichia coli (E. coli) is the most widely used expression system (~30%) because it offers high density fermentations and scale-up production at a minimum cost (1). In terms of scale, productivity and optimal protein folding, and downstream processing, cytoplasmic and periplasmic expression are the sought after means of production (2–4). However, it should be noted that an unbalanced equilibrium between protein aggregation and solubilization results in inclusion body (IB) formation, protein toxicity and inactivity, and low titers of target proteins (5,6).
The product in E. coli is expressed either in cytoplasm or periplasm, or is directly secreted in culture medium (5). The choice of the site of expression depends on a number of factors that include ease of refolding, the role of disulfide bonds on the biological activity of the target proteins, and whether the target protein performs any detrimental and undesirable function in the host cell that interferes with the normal proliferation and homeostasis of the microorganism (7–9). Note that in E. coli, the cytoplasm has a more negative redox potential and this reducing environment is maintained by the thioredoxin–thioredoxin reductase (trxB) and the glutaredoxin–glutaredoxin reductase (gor) system (10). Any disulfide bond formation in the cytoplasm often leads to protein inactivation, misfolding, and aggregation. In contrast, myriad enzymes particularly from the Dsb family catalyze the cysteine oxidation via disulfide exchange reactions in the periplasm where a naturally oxidizing environment is present (11). The advantages and disadvantages of the different sites of protein localization are summarized in Table I. The site of protein localization significantly impacts the consideration of choosing the site of expression of recombinant proteins. Often the decision is guided by several factors including the importance of disulfide bonds for the activity of the protein, if the protein can easily be refolded into its native three-dimensional (3D) form in vitro where the protein expression is in the form of IBs, and if any instances of protein toxicity to the host organism are observed.


After the subcellular location for protein expression has been decided, the next challenge is the availability of an appropriate analytical method that can measure the concentration of the target analyte in complex matrices that fermentation broth usually offers (12). In addition, since bioprocessing of protein therapeutics involves multivariate interactions among feed materials, process variables, and product attributes, it is important to screen as many product attributes as possible with regard to the input variables for obtaining an optimal production strain (13). Specifically, in the case of antibody fragments, the product concentration in the fermentation broth is quite low and in addition there is a heterogeneous population of the product in the broth. When identifying the analytical method of choice, desirable attributes of the method include accuracy, sensitivity, dynamic range, reproducibility, time-to-results, cost, and throughput (14).
Commonly used analytical methods that are employed for antibody fragment quantification include enzyme-linked immunosorbent assay (ELISA), electrophoretic techniques such as sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), immunospecific methods such as western blotting, and chromatographic techniques such as reversed-phase high performance liquid chromatography (HPLC). More recently, the use of optical techniques measuring biomolecular interactions (biolayer interferometry [BLI] and surface plasmon resonance [SPR]) have been explored by researchers (15). Among these, ELISA is the most widely used technique for quantification, but these techniques have limited accuracy especially in the case of complex mixtures like culture broth and crude extracts (16). Also, these techniques cannot discriminate amongst the molecular variants of the product (for example, mass and charge isoforms) versus its native form. Chromatographic quantification methods are well proven and widely accepted, but require laborious and time intensive sample pretreatment and analysis, in addition to requiring significant capital investment for equipment.
Biosensor-based fragment screening is gradually becoming an established practice in drug discovery (17). BLI and SPR spectroscopy are being increasingly used for this purpose (18). Unlike SPR, BLI does not rely on samples flowing through microfluidics, and up to 8–16 protein-labeled sensors can be simultaneously dipped directly into different solutions of small molecules arrayed in a 96- or 384-well plate.
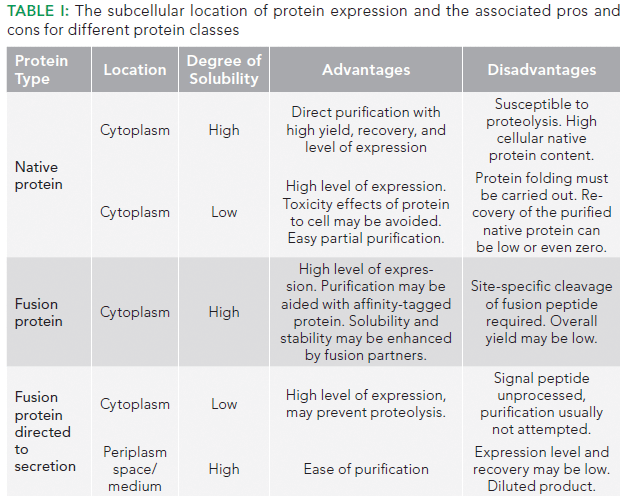
In this installment, we showcase the use of BLI for measuring titer of a product: a fragment antigen-binding (Fab) fragment that has been expressed in periplasm of the E. coli. HPLC based quantification is also performed to showcase the high throughput characteristics of BLI versus HPLC. The screening workflow of sample analysis by BLI and HPLC is illustrated in Figure 1.

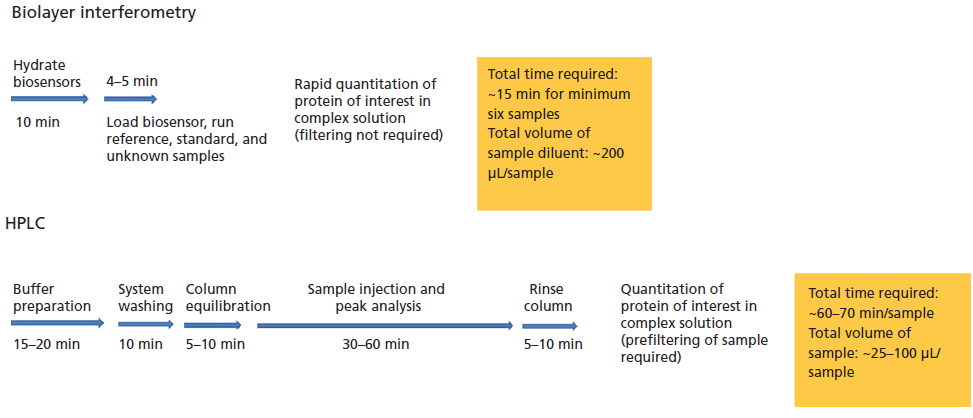
Figure 1: Illustration depicting the screening workflow in the BLI system and the HPLC system.
Theory
Biolayer Interferometry
BLI is a label-free optical analytical technique for measuring molecular interactions between proteins, peptides, and other molecules in a high-throughput manner using nanomole amounts of samples. The method relies on developing an interference pattern from constructive and destructive waves of a reference layer compared with the reflection at the biolayer (Figure 2). The interference pattern is in terms of the wavelength shift proportionate to the changes in thickness as well as local refractive index changes located at the end of the biolayer interferometry biosensor tip as a result of the molecule binding to the biolayer. The shift in the wavelength is in turn proportional to the number and mass of molecules binding on the biosensor tip. This value is recorded as Δnm or Δλ. The small sample requirements make this technique an excellent choice for the analysis of proteins that are otherwise challenging to isolate. In addition, it is also possible to carry out experiments in parallel making this technique suitable for high-throughput analysis of a large number of samples.

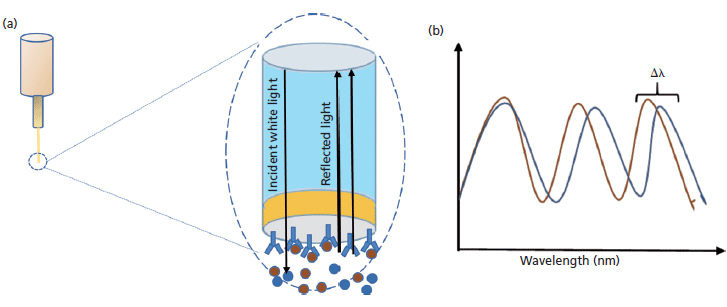
Figure 2: (a) Schematic of BLI biosensor tip, and (b) the wavelength shift.
Another feature of BLI that is relevant to the analysis of biopharmaceuticals is that the refractive index changes in the sample do not affect shifts in the interference pattern (19). This lack of effect is due to the property of the BLI signal detection, which occurs in response to interactions at the tip of the biosensor, and any changes to the matrix and unbound proteins in solution (typically found in crude lysates) have a minimal effect on the signal. This property enables BLI to quantify protein concentrations from heterogeneous crude lysates, thereby circumventing the need for extensive sample preparation steps involving purification and dilution. This advantage allows for significant reduction in the analysis time with significantly high accuracy and precision when compared to traditionally used alternatives. Also, appropriate subtraction methods during data analysis can be effectively used to alleviate nonspecific interactions.
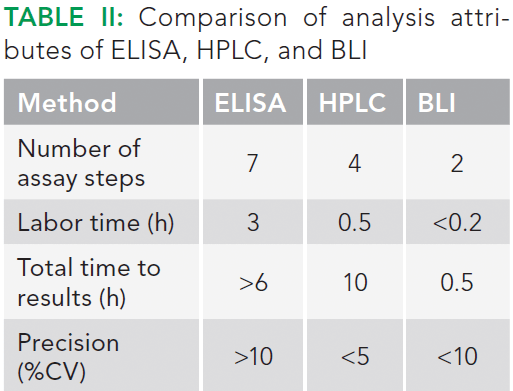
Various biosensor choices based on different chemistries are available that enable quantitation of the proteins. These include anti-human IgG Fc, anti-murine IgG Fv, protein A, protein G, protein L, anti-penta-HIS, streptavidin, anti-human Fab-CH1, anti-GST, and Ni-NTA based biosensors (20). All of these chemistries have very high specificity to the target analyte and can measure in the presence of complex host cell proteins and other components, thereby simplifying analysis at all stages of bioprocess development. In contrast, the traditional techniques for quantitation such as ELISA and HPLC require extensive labor input, sample preparation, and longer time to results. A comparison of these attributes of BLI, ELISA, and HPLC is shown in Table II.

Several key applications of BLI in pharmaceutical development have emerged recently. These include identification of antibody candidates with promising affinities and dissociation kinetics, screening of crude hybridoma supernatants, libraries to identify clones with high affinities and low off-rates, and secondary screening for scFv, Fab, and other biologics from phage or yeast display libraries. Additionally, BLI has been used to characterize antibodies that have undergone affinity maturation, thereby guiding the iterative evolution of antibody hits into lead candidates.
Principle of Fab Quantification by BLI
Protein L possesses a high affinity for the kappa light chain of antibodies and antibody fragments (21). The assay involves coating the tip of the biosensor with a special optical layer followed by capturing molecules (protein L) on this tip (Figure 3). The tip is then dipped into the sample containing the target molecule. The target molecules, upon binding to the captured molecule, form a molecular layer. A white light directed to this assembly gets reflected into two beams. The first beam comes from the tip as a reference while the second light comes from the molecular layer. The difference of the two beams results in the formation of a spectrum color pattern, as depicted in Figure 2b. The phase is a function of the molecular layer thickness and corresponds to the number of molecules on the tip surface.

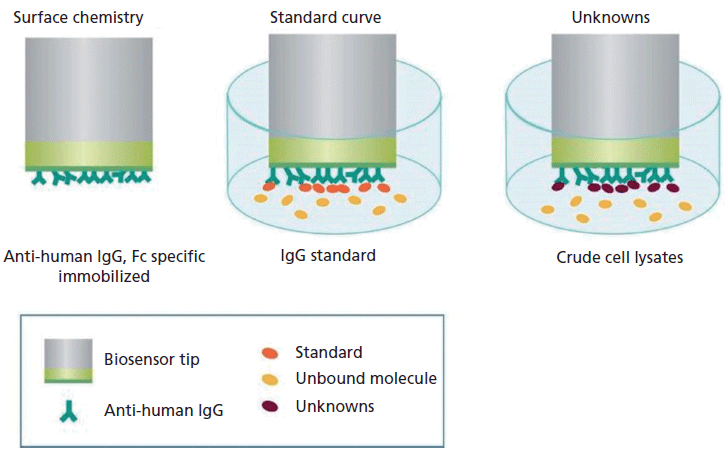
Figure 3: Depiction of the principle of how BLI is used for quantitation of target proteins from a complex matrix.
Materials and Methods
Cultivation
The therapeutic antibody fragment used in this study was expressed in E. coli BL21 (DE3) using rhamnose promoter (pRha). Bacterial cells were cultivated in chemically defined R9 medium containing kanamycin (30 µg/mL) at 30 °C and 200 rpm (22). Protein expression was induced by the addition of 50 mM rhamnose at an OD600 of ~6 and the protein was extracted from periplasm by osmotic shock treatment.
Fab Quantification by Reversed Phase-HPLC
Samples were analyzed by reversed-phase HPLC on an Ultimate 3000 HPLC system (Dionex, Thermo Scientific) using a 150 mm × 4.6 mm Zodiacsil column operated at 70 °C with a flow rate of 0.5 mL/min. Mobile-phase A was composed of 0.1% trifluoroacetic acid in purified water (MilliQ, EMD Millipore) and mobile-phase B was composed of 0.1% trifluoroacetic acid in acetonitrile. The equilibration of the column was achieved using 45% B for 5 min. The elution was performed using a linear gradient of 45–100% B in 25 min. The column was regenerated by using 45% B for 15 min. Protein detection was performed by ultraviolet (UV) absorption at 214 nm.
Fab Quantification by BLI
An Octet RED96 BLI system (Fortebio, Pall) was used for quantification of the antibody fragment using protein L biosensors (ForteBio). Black 96-well microplates were obtained from Greiner Bio-One. A commercial standard of the therapeutic antibody was used as a calibration standard. All the samples analyzed were diluted in sample diluent (ForteBio), consisting of 10 mM PBS along with 0.1% BSA and 0.02% Tween 20, pH 7.4.
Assay Protocol
Preparation of Samples and Calibration Standards
The samples, calibration standards, and hydration solution in the sensor plate (200 µL/well) were prepared as per the procedure illustrated in Figures 4a and 4b. Hydration of the protein L sensor was performed in phosphate-buffered saline (PBS), pH 7.4. The regeneration was performed in 10 mM glycine, pH 1.5, and the neutralization buffer was kept identical to the hydration buffer. Since exposure of hydrated biosensors to air may affect performance, the hydrated biosensors were kept ready for the binding experiments with the analyte. Biosensors hydrated in the BLI kinetics buffer retain activity when stored overnight in the BLI kinetics buffer at 4 °C.

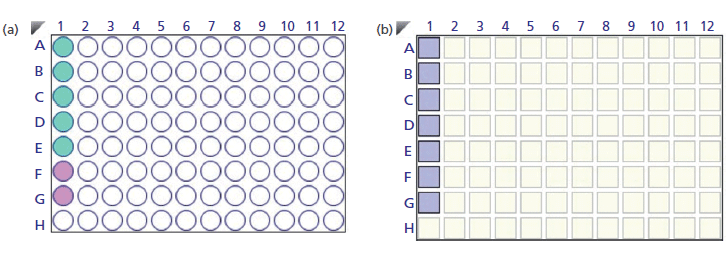
Figure 4: (a) Layout of standard solution (green) and unknown samples (purple) in the sample plate. (b) Layout depicting the position of protein L sensors in the sensor plate.
Preparation of Assay Plate and Biosensors
The control and assay samples were pipetted (200 µL/well) and the hydration solution was placed in the wells corresponding to the position of the biosensor to be used in the analysis. Next, they were hydrated for 10 min before the experiment followed by sample measurement at 1000 rpm for 300 s. Alternatively, the delay timer can be used to automatically start the assay after 10 min. All experiments were performed at 30 °C. The assay was performed in the basic quantitation with regeneration assay format of the Octet data acquisition software.
Data Analysis
Data analysis was performed using Octet data analysis software version 7.1. The acquired data were corrected by subtracting the black matrix from the sample matrix. The reference subtracted data was analyzed using the initial slope binding rate equation.
Results and Discussion
Fab Quantification Using HPLC
Reversed-phase HPLC plays a critical role with respect to characterization of monoclonal antibodies (mAbs). These products include full-sized intact mAb, Fab, and Fc fragment molecules, reduced heavy- and light-chain species, and peptide maps generated by proteolytic digestion. There are many reasons that make reversed-phase HPLC a popular tool, including the well-studied hydrophobic separation mechanism, the availability of efficient reversed-phase-HPLC columns packed with small-particle, nonporous, fully porous, or superficially porous (SPP) materials, and the use of mobile phases compatible with mass spectrometry (MS) for peak identification and structure elucidation.
Figures 5a and 5b illustrate the chromatograms of the standard molecule and the target protein (Fab protein) obtained using reversed-phase-HPLC. The quantification was done by comparing the peak area of the standard protein (known concentration) to the peak area of the target protein in the periplasm extract (prefiltered) based on identical retention times. Protein samples were concentrated by using 10-kDa cut-off centrifugal filtration and retentate. In addition, filtrate proteins were analyzed for estimation of the Fab molecule.

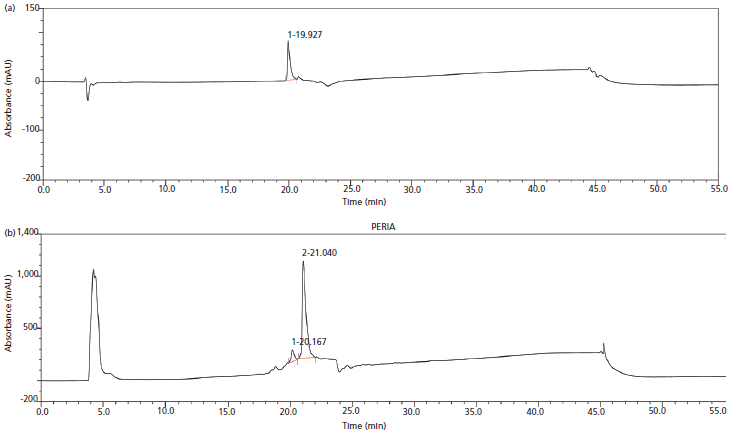
Figure 5: (a) Reversed-phase-HPLC chromatogram of standard molecule. (b) Reversed-phase-HPLC chromatogram of Fab molecule expressed in the periplasmic compartment of E. coli.
As indicated in Table II, the advantage that reversed-phase HPLC offers is the high resolution for separation of antibody fragments. The analysis time is about 45 min per sample. In addition, every consecutive sample injection requires intermittent blank runs to prevent carryover. Thus, in cases for which a large number of samples are to be analyzed, such as drug discovery or during process development, HPLC analysis is often the bottleneck.
Fab Quantification Using BLI
First, standard curves were constructed using a purified Fab molecule at concentrations of 5, 10, 15, and 25 µg/mL. Standard curves were assayed in different well locations in three independent experiments to account for any well-to-well variability. A consistent assay performance across all the wells was observed with average coefficient of variation (CV) for the entire standard curve range of less than 11.5%.
Next, possible interference of the media components with the analysis was assessed by constructing standard curves using various dilutions of the culture media and then overlaying and assessing the degree of variability at each of the concentrations. The assay was found to tolerate up to 100% culture media, with comparable recoveries of spiked standards in all dilutions and an overall CV of <10% (Figure 6). These results indicate that BLI offers a robust method for analysis of upstream process samples with minimal preprocessing.

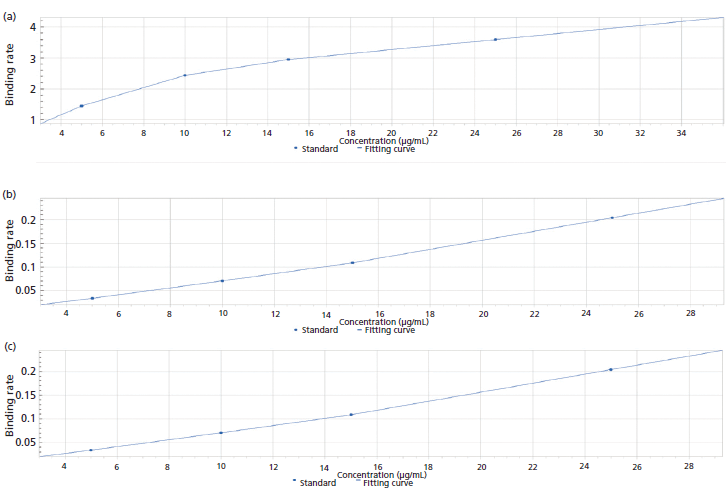
Figure 6: Standard curve generated using various dilutions of culture media: (a) neat; (b), twofold, 50% culture medium; (c) fivefold, 20% culture medium.
Figures 7a–7c represent the binding curve generated for the standard Fab molecule. The binding rates of the test sample were measured and interpolated from the standard curve to determine the concentration. Data for eight wells were acquired every 2 min, thereby enabling analysis of 96 samples in ~30 min. Significantly, there are no wash steps required during analysis from one well to the other. It should be noted that the use of a standard curve for determining the concentration of the test sample is optional; however, it is recommended that separate standard curves be constructed for different culture broths. The reported values of the assay sensitivity for Fab fragment quantification were 0.05–300 µg/mL. For the present study, the dynamic range was found to be 100–300 µg/mL.

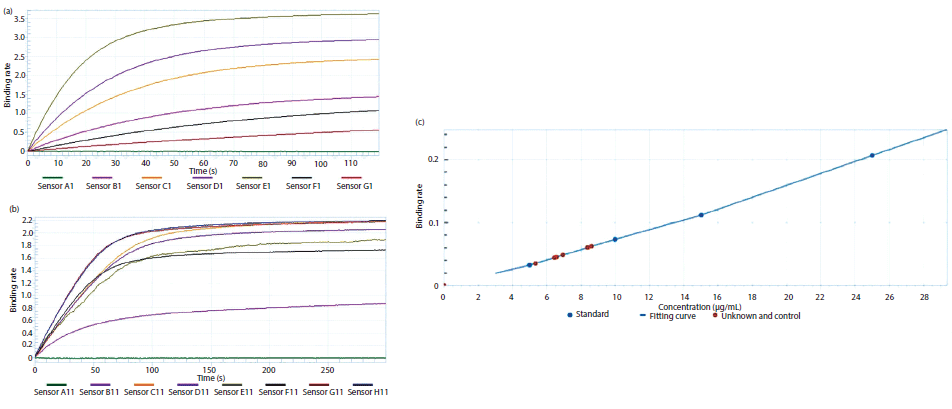
Figure 7: Real-time binding data of (a) standard Fab molecule and (b) expressed Fab molecule, and (c) a graph depicting the binding rate of standard as well as target Fab protein.
Comparison of Titer Measurement by HPLC and BLI
A comparison of the performance of HPLC and BLI to quantify the Fab fragment was performed using seven periplasmic-expressed Fab proteins. For BLI, the samples were diluted 40-fold so that the concentration was brought within the dynamic range of the instrument. For HPLC, the analysis was performed by injecting 100 µL of the undiluted samples. There was no other difference in the sample preparation strategy for the analysis using these two platforms; the goal was to obtain an unbiased comparison of the two methods.
Titer values measured with both techniques showed reasonable comparability with a deviation of ~13% between the two techniques (Table III). However, for most of the samples, the differences between the values obtained using the two techniques were well within 10%.

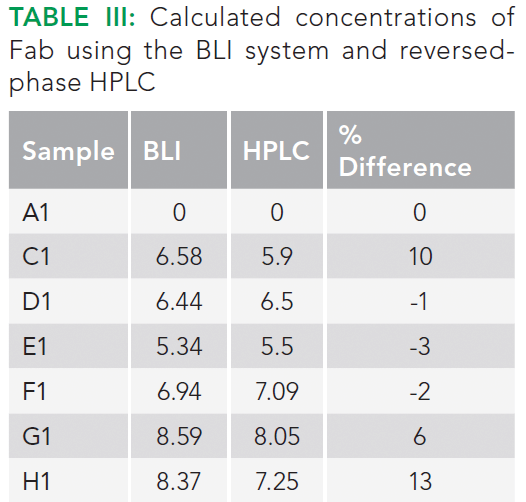
With regards to the total analysis time, it is clear that BLI offers a significantly faster analysis time (0.5 h versus 90 h) for 96 samples. The time includes any intermediate processing steps that may be required during analysis using both techniques. Note that the injection of crude sample will reduce the column life; 200 injections of crude samples versus 1000 injections of clean samples. In contrast, BLI does not suffer from these shortcomings.
Reversed-phase HPLC, however, can provide information about the presence of various molecular variants and other product heterogeneities of the sample. BLI is unable to provide this information. Thus, the two techniques complement each other and together they can be used for a comprehensive quantitation and analysis of Fab fragments in complex culture media. Table IV compares the various characteristics of sample analysis by HPLC and BLI.

Limitations of Affinity Recognition–Based Approaches
There are, however, a few generic deficiencies in any analytical assay that utilizes affinity recognition principles to detect or identify the target protein or antibody. These deficiencies have been known for decades, ever since ELISA was discovered and promulgated around the world, to the present day. First off, molecular recognition entities, especially antibodies (mAbs), proteins, or peptides, are not always easy to manufacture in a totally reproducible manner, batch to batch, even for analytical reagent purposes. The current dilemma in the scientific literature related to a general inability to reproduce certain publications using antibodies, such as ELISA, has been attributed to the difficulties in obtaining identical mAbs or proteins from the same vendor, year to year, or from different vendors at any given time. This inconsistency then leads to a general, at times, lack of reproducibility of the original studies, unless the authors had done a thorough analytical method validation demonstration in their original publication (not usually the case today).
A second, generic problem in using affinity recognition is that very few such reagents recognize only the analyte molecule of interest in developing the original assay, whatever that might have been. Biological routes to recognition molecules rarely evolve any protein that will recognize only a single, molecular entity. This drawback has been known from the very beginning of ELISA, which also should not be used alone or entirely for identifying any specific antigenic species until it is shown to be 100% specific. That is almost never the case, nor will it be for the recognition entities in BLI.
There is a third possible problem with using BLI alone to identify the precise structure of any protein: not knowing if that protein is alone at the biorecognition step. BLI does not resolve all the entities that might be present in the final sample under analysis-it is not chromatography or electrophoresis and has no separation powers beyond affinity recognition, by and large.
Finally, BLI does not provide any indication of the molecular weight or structure of the antigenic species under analysis. Though it may be used for approximate quantitation, it does not truly identify the analyte species, other than that it recognizes it by some form of molecular interaction processes. It provides no structural or molecular weight information, no fragmentation data, and no database that could be approached to confirm its recognition by molecular interactions or fragmentation patterns, which are so common in MS methods of analysis and quantitation today.

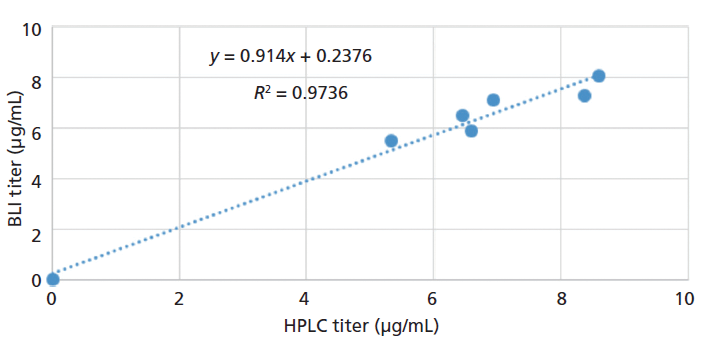
Figure 8: Comparison of Fab measured by BLI and reversed-phase HPLC methods.
Conclusions
Despite the caveats discussed above, it should be noted that affinity recognition is playing an increasingly significant role in many protein analysis methods now being developed, especially in bioanalysis. BLI can be considered a rapid, inexpensive, and fully automatable approach for affinity recognition, as in ELISA, and partial qualitative identification of a protein in a complex sample or biofluid, along with suitable quantitation having authentic reference standards run alongside the actual samples of interest. This study amply demonstrates the utility of BLI as an alternative to HPLC for quantitation of protein samples in complex media matrices. The utility is particularly more pronounced where there is a demand for high-throughput analysis without affecting sensitivity and robustness of analysis. The ability of protein L to retain its binding activity ensures that the biosensors can be regenerated and the necessary optimization of conditions would maximize the recovery.
Acknowledgments
This work was funded by the Center of Excellence for Biopharmaceutical Technology grant from Department of Biotechnology, Government of India (number BT/COE/34/SP15097/2015). The authors greatly acknowledge Susheelendra Vaidya and Sriram Kumaraswamy from ForteBio, a Division of Pall Life Sciences for their help with Octet.
References
(1) R. Chen, Biotechnol. Adv. 30, 1102–1107 (2012).
(2) G.P.L. Cereghino, J.L. Cereghino, C. Ilgen, and J.M. Cregg, Curr. Opin. Biotechnol. 13, 329–332 (2002).
(3) A.L. Demain and P. Vaishnav, Biotechnol. Adv. 27, 297–306 (2009).
(4) S. Hellwig, J. Drossard, R.M. Twyman, and R. Fischer, Nat. Biotechnol. 22, 1415–1422 (2004).
(5) F. Baneyx, Curr. Opin. Biotechnol. 10, 411–421 (1999).
(6) G.L. Rosano and E.A. Ceccarelli, Recomb. Protein Expr. Microb. Syst. 7, 172 (2014).
(7) H. Dong, L. Nilsson, and C.G. Kurland, J. Bacteriol. 177, 1497–1504 (1995).
(8) A.I. Derman, W.A. Prinz, D. Belin, and J. Beckwith, Science 262, 1744–1748 (1993).
(9) A. Basu, X. Li, and S.S. Leong, J. Appl. Microbiol. Biotechnol. 92, 241 (2011).
(10) E.J. Stewart, F. Åslund, and J. Beckwith, EMBO J. 17, 5543–5550 (1998).
(11) J. Messens and J.-F. Collet, Int. J. Biochem. Cell Biol. 38, 1050–1062 (2006).
(12) M. Pathak, S. Dixit, S. Muthukumar, and A.S. Rathore, J. Pharm. Biomed. Anal. 126, 124–131 (2016).
(13) S.M. Mercier, B. Diepenbroek, M.C.F. Dalm, M. R.H. Wijffels, and M. Streefland, J. Biotechnol. 167, 262–270 (2013).
(14) S.M. Mercier et al., Eng. Life Sci. 16, 25–35 (2016).
(15) M.E. Davies, A.J. Barrett, and R.M. Hembry, J. Immunol. Methods 21, 305–315 (1978).
(16) H. Lei, W. Chong, Q. Wei-zhu, and W. Hao, Curr. Immunol. 4, 4 (2012).
(17) D.E. Scott, A.G. Coyne, S.A. Hudson, and C. Abell, Biochemistry 51, 4990–5003 (2012).
(18) S. Gao, X. Zheng, and J. Wu, Sensors Actuators B Chem. 246, 169–174 (2017).
(19) "Rapid , Reliable Quantitation of Fc-Fusion Protein in Cell Culture Supernatants," available at: http://www.fortebio.com/.
(20) ForteBio, http://www.fortebio.com/.
(21) B.H. Nilson et al., J. Immunol. Methods 164, 33–40 (1993).
(22) D. Riesenberg et al., J. Biotechnol. 20, 17–27 (1991).
ABOUT THE AUTHORS

Deepak Kumar

Deepak Kumar is a post-doctoral fellow in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.

Jyoti Batra

Jyoti Batra is a research scientist at Vallabhbhai Patel Chest Institute, University of Delhi, India.

Anurag S. Rathore

Anurag S. Rathore is a research scientist at Vallabhbhai Patel Chest Institute, University of Delhi, India.

Ira S. Krull

Ira S. Krull is a research scientist at Vallabhbhai Patel Chest Institute, University of Delhi, India.








Analytical Methods to Determine the Stability of Biopharmaceutical Products
January 1st 2023A drug stability program is a fundamental part of ensuring product quality, safety, and efficacy. Here, we summarize essential guidelines, differences between large- and small-molecule stability, and the analytical methods used.

")
Analytical Characterization of Host Cell Proteins (HCPs)
October 1st 2022In response to regulatory concerns, host cell protein (HCP) analysis is now often conducted using LC–MS/MS. Unlike ELISA, LC–MS/MS can positively identify and quantify specific HCPs and characterize the total amount of HCPs present.

, also known as an immunoglobulin (Ig). 3d vector.")

