Bare Silica Has a Future in Supercritical Fluid Chromatography
LCGC Europe
SFC is now regarded as a modern normal phase LC technique prompting a re-examination of the role of bare silica as a useful stationary phase.
The realization that supercritical fluid chromatography (SFC) is now viewed as modern normal phase (NP) liquid chromatography (LC) has prompted a re-examination of bare silica as a viable stationary phase for packed column SFC. Published documents that appeared before and after 2000 involving bare silica and CO2/methanol mobile phases are reviewed. A current study which experimentally compares five bonded phases with bare silica for the SFC separation of a 19-component mixture of polar analytes is reported.
The evolution of stationary phases in supercritical fluid chromatography (SFC) continues to take place, even though the technique is now over 50 years old. Initially packed columns were used for SFC in the 1960s, but in 1981 attention switched to cross-linked polymer wall coated open tubular columns (WCOT). The most popular stationary phases (SP) were siloxane-based aliphatic and/or aromatic hydrocarbons that were analogous to (at the time) popular gas chromatographic phases. SFC open tubular columns, however, had smaller inner diameters than conventional GC columns. Packed columns (PCs) were regarded as being too retentive due to the active nature of the packing, although Berger insisted that the retentiveness was solely due to differences in the "phase ratio" of packed versus open tubular columns (OTCs).1 The controversy quickly died as it became clear that the two techniques (i.e., OTC versus PC) tended to best address different problems.
Nowadays, the future of SFC is considered to rest unmistakably with packed, bonded silica-based columns and carbon dioxide-based mobile phases (MPs). The early goal of using only pure carbon dioxide was, however, realized only in part with nonpolar and weakly polar analytes. The limited success was attributed to many unsuccessful attempts to deactivate silica-based packing materials.2 The obvious strategy of conventional endcapping for each HPLC phases was not effective in that all silanol sites were not reacted and at temperatures above 100 °C the endcapped reagent was not stable.
For a short while a more satisfactory solution to this dilemma involved the use of hydrosiloxane polymers that were coated and chemically bonded to the porous silica particle. A surface coating technique similar to one previously developed for a capillary GC column was used. The bonded phase would ideally cap or shield any remaining active sites from involvement in the separation mechanism. Results from this approach or its modification were surprisingly successful; resulting in the elution of moderately polar aliphatic amines with 100% CO2 without an additive or even a modifier.3 Nevertheless, it became apparent to workers in the field that binary phases of CO2 and an organic polar modifier such as methanol would be required for elution of highly polar analytes that would be of interest to the pharmaceutical industry.
SFC is now a separation technique most similar to normal phase HPLC. It almost always uses carbon dioxide as the main component in a binary or ternary mobile phase. It is usually performed as gradient elution where the composition of the mobile phase is changed versus time. Pressure programming has become uncommon. Small changes in temperature are used to change the selectivity of difficult analyte pairs. While SFC is quite amenable to the separation of volatile, non-polar analytes, it usually does not compete very favourably with gas chromatography (GC). The future of SFC is, therefore, the separation of highly polar analytes, which was the case many years ago with normal phase liquid chromatography (NPLC). SFC is now more applicable, more efficient, faster, greener and scalable for preparative chromatography than conventional NPLC.4 Considered by most workers in the field to be modern NP chromatography, SFC generally requires polar stationary phases to separate most polar solutes. In this manuscript, no differentiation will be made here between what is sometimes called subcritical fluid chromatography, enhanced fluidity chromatography and SFC. At sufficiently high pressure, transitions between these pseudo states are undetectable chromatographically and the instrumentation used is identical.
Stationary Phase Modification
HPLC studies have shown that a monolayer of the strongest solvent component of the mobile phase commonly exists on the surface of the stationary phase. Comparable studies have shown that somewhat similar effects are observed for SFC with CO2 as the mobile phase. Adsorption isotherms for supercritical CO2 displayed maximum adsorption at pressures close to the critical pressure with silica, octadecyl-, cyano- and diol-adsorbents.5 Multilayer adsorption occurred in each system and the type of bonded phase had little or no effect on the adsorption. In other words, under conditions normally used for packed column SFC, significant amounts of not only CO2 but also modifier are adsorbed on the solid adsorbent and function as components of the stationary phase.



KEY POINTS
Parcher has thus suggested that polar modifiers can possibly affect the performance of an SFC system in a variety of ways.6 For example, (a) the modifier may alter the density and solvent power of the MP, (b) the modifier can block active adsorption sites on the SP inhibiting adsorption of solutes, (c) sorbed modifier may act as a component of the SP, (d) sorbed modifier may increase the volume of the SP while decreasing the volume of the MP thereby changing the phase ratio for the column and (e) the modifier may also solvate around a polar solute to form clusters in the MP. The combination of all these factors could result in either enhanced or diminished retention of solutes with variations in modifier concentration. All five effects may operate in any given SFC procedure that uses subcritical, polar modifiers. It is safe to assume, nevertheless, that an important aspect in the use of modifiers, besides changing the density and solvating power of the MP, is the influence of the modifiers and supercritical fluids on the physical and chemical properties of the SP. The effects of temperature, pressure and composition on the adsorption of both methanol and CO2 have been determined over a range of conditions with supercritical fluid MP. Under the conditions normally used for packed-column SFC, significant amounts of CO2 and modifier were both adsorbed on the solid adsorbent and functioned as components of the SP.
Many acidic, basic and amphoteric compounds do not elute or elute with poor peak shapes even when the mobile phase consists of a binary mixture of CO2 and a modifier such as methanol and acetonitrile. The addition of a very polar secondary modifier (i.e., additive) appears to allow elution of symmetrical peaks.7 Just as with primary modifiers, additives may (a) suppress solute ionization, (b) adsorb on active sites of the SP and (c) form neutral ion pairs with ionic solutes. Additives adsorb onto many, but not all stationary phases. Surface coverage between 0.4 and 21% was measured at millimolar or lower mobile phase concentrations. The amount adsorbed increased with increasing stationary phase polarity. Changing concentration of the modifier appeared to affect the amount of additive adsorbed. The higher the concentration of modifier, the smaller the amount of additive adsorbed.8
In light of the observation that all components of the polar modified MP in supercritical fluid chromatography are sorbed to different degrees on the SP, one might question the nature of the SP that was purchased versus the nature of the SP that is actually being used for separation. In other words, would a bare silica column suffice just as well as a bonded silica phase for SFC because mobile phase components are modifying the nature of the polar bonded phase silica? With the advent of purer and better defined silica that contains less metal ion contamination by major vendors, the question seems worth asking. Because silica columns appear to be more economical today than bonded phases, the question takes on added significance, especially for preparative chromatography. A recent review of the published literature has suggested that bare silica continues to find increased use in packed column SFC.
Early SFC Studies with Bare Silica
Silica serves as the solid support for many stationary phases, but historically bare silica has been most often used as the stationary phase for absorption liquid chromatography (i.e., normal phase). In their pioneering SFC work, Klesper et al. mentioned having tried silica gel in different publications. Soon after this early work spherical porous silica particles became available that provided more uniform shape, higher surface area and greater sample capacity that packed more efficiently. Particle sizes varied from 3–10 μm in diameter with pore sizes ranging from 60 to 300 angstroms corresponding to a surface area of approximately 100 to 300 m2 g–1. The surface activity of silica packing materials proved to be a serious limitation when CO2 was used to elute moderately polar analytes. Chromatographic results, however, became markedly different when binary and ternary mobile phases were used with silica.
In 1986 the influence of the physical state of CO2 concerning solute retention on bare silica was studied.9 The retention mechanism in SFC with CO2 was found to be very similar to that observed in HPLC with an apolar solvent such as hexane. SFC retention decreased when pressure (i.e., density) increased at constant temperature. However, when temperature was increased at constant pressure with phenanthrene and naphthalene as analytes, two situations were encountered. Below 200 bar, retention increased as solubility decreased, which was to be expected. Above 200 bar, solubility increased with temperature but retention surprisingly increased also. This increase in solute–stationary phase interaction could only be explained by a change in the apparent nature of silica caused by the presence of water which could affect SP activity. Water solubility in SF CO2 with temperature at 200 bar. For example, at 25 °C, solubility is 1500 ppm; while at 75 °C, solubility is 4500 ppm. Thus, when temperature increases, some water molecules are transferred from the SP to the MP and the number of free silanol groups covered by water molecules decreases. The adsorption energy of the solutes on the free silanol sites should be higher; thus there can be a simultaneous increase in retention and solubility.
After this study, non-bonded silica was observed to give a higher resolution than bonded aminopropylsilica for the analysis of narcotine, thebaine, codeine, cryptopine and morphine.10 The mobile phase was a CO2/methanol/methylamine/water mixture (83.37:16.25:0.15:0.23). The procedure was later applied to a poppy straw extract and to the determination of three alkaloids.
In another early study, capacity ratios and selectivity were determined for basic compounds, fat-soluble vitamins and carboxylic acids.11 Retention behaviour of the model compounds was compared for SP of unmodified and chemically modified silica particles. Unmodified silica gave higher plate numbers for more of the test solutes than the bonded stationary phases. Therefore, pure silica was recommended when the substances of interest could be eluted with reasonable capacity ratios and adequate selectivity.
The separation and identification of sesquiterpene hydrocarbon mixtures on bare silica as a stationary phase with Fourier transform infrared detection and 100% CO2 was also reported approximately 20 years ago.12 The separation of these nonpolar but extremely thermosensitive organic compounds required low temperature rather than low density. These chromatographic conditions preserved structural information about the sesquiterpene hydrocarbons such as the differentiation of methyl and methylene groups and the geminal or single nature of methyl groups. In a related investigation, bare silica stationary phase with a high specific surface area was considered to give higher selectivity than a partitioning SP because of the similar chemical structures of the analytes.13 PCSFC with 0.5% methanol was found to be useful for the separation (10 min) of thermosensitive sesquiterpene alcohols such as nerolidol, alpha-bisbolol, farnesol and geranyllinalol. The elution order on silica was the same as observed in adsorption HPLC with 2,3,4-trimethylethane-ether mixtures.
The application of SFC with 10% methanol-modified CO2 for the separation of polymethoxylated flavones (PMF) has been reported.14 Bare silica with UV detection (313 nm) was used. Selectivity was found to be different with SFC than with HPLC. Sinensetin, nobiletin, taneretin, heptamethoxylflavone and tetramethylscutellarein could be quantitatively determined using an internal standard (IS). The method was applied to the determination of PMFs in several citrus oils. SFC has also been more recently reported as a method of analysis for the determination of 4-hydroxybenzylglucosinolate degradation products.15
Recent SFC Studies with Bare Silica
The use of bare silica has been reported for the separation of ionic analytes such as sodium aryl sulphonates with ionic additive and polar modifier in the MP.16,17 The effect of additive concentration and additive functionality on analyte retention was investigated. The additives were lithium acetate, ammonium acetate, tetramethylammonium acetate and ammonium chloride dissolved in methanol. While the positive effect of ammonium acetate was expected from previous reports, results for the other additives salts were not expected and suggested a general phenomenon that seemed to not be cation or anion specific. A similar effect was also observed on the two cyanopropyl bonded phases that were investigated in this work. The fact that bare silica yielded analogous results to the two bonded phases suggested that some alteration of the SP by the additive was strategically involved. Interestingly, the aryl sulphonates were found to be retained more strongly on the silica column when the concentration of ammonium acetate in methanol was increased, which was inverse of the trend with the bonded phases. This observation suggested that different elution mechanisms may be operating with bare silica and the bonded phase silica. The elution mechanism envisioned with bare silica was suggested to be ion exchange; whereas an ion-pairing mechanism was postulated for the bonded phases. The elution of the totally ionic analytes with a CO2-based MP from a highly active, non-bonded silica packed column SP was not predicted and was deemed noteworthy.
Bare silica along with zirconia and titania have been compared for their ability to separate diesel samples into saturates, mono-, di-, tri- and polyaromatics by SFC using pure CO2 according to ASTM method D 5186-03.18 Conventional Type A bare silica column HGS, Lichrospher Si 60, Lichrosorb Si 60, as well as a low metals Type B silica Kromasil Si column were studied to determine how well bare silica columns performed group-type separations of diesel samples. None of the silica columns adequately separated all of the model compounds according to group type, with the separation of the di- and tri-aromatics proving to be the most difficult. Yet, all of the columns studied met the ASTM D 5186-03 method requirements. A titania column coupled in series to a silica column was found to provide the highest overall group-type resolution based on 20 model compounds with resolutions as high as 14.7 for saturates versus monoaromatics and 11.8 for monoaromatics versus diaromatics. An oil sands-derived Synfuel light diesel, a commercial Ontario diesel and a heavy Shell Canada Ltd diesel blending feedstock were studied on both the bare silica column and the coupled columns. The Synfuel results were similar, but the heavier commercial diesel and diesel blending feedstock samples were different.
The separation of cytarabine (ara-C) from the endogenous compounds in mouse plasma by packed column SFC with an isocratic MP composed of CO2/MeOH and ammonium acetate as an additive has recently been reported.19 The packed column was integrated with a tandem APCI-MS–MS to enhance sensitivity, selectivity and speed of analysis. No change in retention for ara-C was observed with an increase of water content in the modifier from 0.01 to 1.0%. The mouse plasma levels of ara-C (ng/mL) were found to be consistent with those determined by various reversed phase, HPLC methods using either a porous graphite carbon column, a mixed-mode column, or a C18 column in conjunction with an ion pairing agent coupled to a tandem mass spectrometer.
Given the relatively few references discussed in this treatise, we feel that this review of packed column SFC with non-bonded silica is nearly exhaustive. The use of bare silica for packed column SFC has been known as long as SFC has been known. Its use, however, has not been embraced by many workers in the area and reported publications have been rather spotty. No doubt silica impurity, traces of uncontrollable mobile phase moisture and a host of newly developed, competing bonded polar phases have led to a lack of interest in silica for SFC purposes. In those instances where it has been used the results appear favourable compared with both HPLC and with SFC that embraces packed bonded silica phases. As the quality of bare silica has improved, no doubt its properties as a stationary phase have become more attractive, especially because water has been demonstrated to be an excellent neutral additive in conjunction with silica and various polar bonded phases.
Current work with Bare Silica
We have experimentally compared five bonded phases with bare silica for the separation of a 19-component mixture of polar analytes (Table 1) with CO2, methanol modifier, and 3% water/10 mM ammonium acetate (AA) as a binary additive (Figure 1). All separations were performed using a SFC instrument (Thar, Pittsburgh, Pennsylvania, USA) equipped with a fluid delivery module, column oven, autosampler, automated back pressure regulator and a 2998 photodiode array detector (Waters, Milford, Massachusetts, USA ). All chemicals were obtained from Sigma-Aldrich Inc. (Saint. Louis, Missouri, USA); while all solvents were HPLC grade from Thermo Fisher Scientific (Pittsburgh, Pennsylvania, USA). Bare silica, 2-ethylpyridine, diol and cyanopropyl packed columns (250 × 4.6 mm, dp = 5 μm, 60 Å) were obtained from Princeton Chromatography (Princeton, New Jersey, USA). Octadecyl and phenyl-hexyl columns of similar dimension were obtained from Phenomenex (Torrence, California, USA). All chromatographic separations were performed at 3.5 mL/min with the following step gradient of mobile phase components: Time zero: 90/10 CO2/modifier; time 1 min: 60/40, time 10 min: 60/40, time 3 min: 90/10, time 5 min: 90/10. Additives were incorporated into the methanol modifier (v/v) before mixing with CO2. CO2 pressure and temperature were 200 atm and 60 °C. The sample solvent was methanol and the injection volume was 5 μL. Two injections separated by approximately 11 min (during which time the column was equilibrated with the mobile phase) were made for each run. The results of the second injection were accepted and are shown here. Water and ammonium acetate proved to be ideal additives for the separation.20 Overall peak shape and selectivity with silica are as good as or better than with any of the remaining five phases using identical chromatographic conditions. The superiority of polar phases for the separation of polar analytes is clearly demonstrated in the set of six chromatograms. Selected, clearly resolved components of the analyte mixture are identified for the silica column in Table 1.

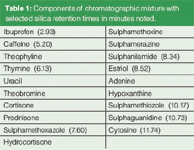
Table 1: Components of chromatographic mixture with selected silica retention times in minutes noted.
In summary, the way to succesfully improve SFC chromatographic peak shapes of polar solutes is by using polar modifiers and even more polar additives with polar stationary phases. The dominant retention mechanism for solutes in SFC then becomes the interaction between the solutes and the most polar entity on the chromatographic support. Berger and Deye suggested that to be successful in SFC the stationary phase should be more polar than the solutes and probably more polar than a silanol site.21 Thus, it is now assumed that a film of modifier and CO2 is a significant, if not dominant part of the true packed column stationary phase because the film tends to further decrease the intensity of the solute—silanol interaction. Under supercritical fluid conditions, surface molar excesses of mobile phase additives may also act to change the chemical nature of the stationary phase surface thereby making it more polar than the mobile phase. The excellent separation of polar analytes on bare silica clearly supports these hypotheses.

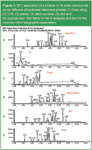
Figure 1
Conclusions
The greatest success for improving SFC chromatographic peak shapes of polar solutes is achieved using polar modifiers and even more polar additives with polar stationary phases. The dominant retention mechanism for solutes in SFC then becomes the interaction between the solutes and the most polar entity on the chromatographic support. Berger and Deye suggested many years ago that to be successful in SFC, the stationary phase should be more polar than the solutes and probably more polar than a silanol site.21 Thus, it is now assumed that a film of modifier and CO2 is a significant, if not dominant part of the true packed column stationary phase because the film tends to further decrease the intensity of the solute—silanol interaction. Under supercritical fluid conditions, surface molar excesses of mobile phase additives may also act to change the chemical nature of the stationary phase surface, thereby making it more polar than the mobile phase. The excellent separation of polar analytes on bare silica clearly supports these hypotheses.
Acknowledgment
The loan of a Waters SFC (Milford, Massachusetts, USA) instrument is greatly appreciated. Financial support of this work was provided in part by Applied Analytical Inc. Discussions with Frank Riley and Jian Wang at Pfizer (Groton, Connecticut, USA) and Rui Chen (Waters) were extremely helpful.
Medhi Ashraf-Khorassani received his Ph.d in Analytical Chemistry from Virginia Tech in 1998. From early 1992 till mid-1994 he joined Trabriat Modaress University (Tehran, Iran) as an assistant professor). In 1994 he joined Virginia Tech as Senior Research Scientist in the chermistry department.
Larry T. Taylor is Emeritus Professor of Chemistry, Virginia Tech. He has served as the Chair of the Scientiific Committee for SFC 2008 (Zurich), SFC 2009 (Philadelphia), and SFC 2010 (Stockholm). He is the author or co-author of approximately 400 peer-reviewed publications and 12 patents.
References
1. T.A. Berger, "Packed Column SFC", RSC Chromatography Monograph Series, Royal Society of Chemistry, Cambridge, Chapter 4 (1995).
2. L.T. Taylor and H-C.K. Chang, J. Chromatogr. Sci., 28, 357–366 (1990).
3. M. Ashraf-Khorassani, L.T. Taylor and R.A. Henry, Anal. Chem., 60, 1529–1533 (1989).
4. L.T. Taylor, J. Supercrit. Fluids, 47, 566–573 (2008).
5. J.F. Parcher and J.R. Strubinger, J. Chromatogr., 479, 251–259 (1989).
6. J.R. Strubinger, H. Song and J.F. Parcher, Anal. Chem., 63, 104–108 (1991).
7. T.A. Berger and J.F. Deye, J. Chromatogr., 547, 377–392 (1991).
8. T.A. Berger, J. Chromatogr., 785, 3–33 (1997).
9. P. Mourier et al., J. Chromatogr., 353, 61–75 (1986).
10. J.L. Janicot, M. Caude and R. Rosset, J. Chromatogr., 437, 351–364 (1988).
11. D. Upnmoor and G. Brunner, Chromatographia, 28, 449–454 (1989).
12. P. Morin et al., J. Chromatogr., 464, 125–137 (1989).
13. P. Morin, Fresenius J. Anal. Chem., 348, 327–328 (1994).
14. P. Morin et al., J. Chromatogr., 586, 171–176 (1991).
15. S. Buskov et al., J. Biochem. Biophys. Methods, 43, 157–174 (2000).
16. J. Zheng et al., J. Chromatogr. A, 1082, 220–229 (2005).
17. J. Zheng et al., J. Chromatogr. A, 1090, 155–164 (2005).
18. R.E. Paproski, J. Cooley and C.A. Lacy, J. Chromatogr. A, 1095, 156–163 (2005).
19. Y. Hsieh, F. Li and C.J.G. Duncan, Anal. Chem., 79, 3856–3861 (2007).
20. M.A. Ashraf-Khorassani and L.T. Taylor, J. Sep. Sci., accepted for publication.
21. T.A. Berger and J.F. Deye, J. Chromatogr. Sci., 29, 280–286 (1991).

")














Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).




Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

