Advances in Liquid Chromatography–Tandem Mass Spectrometry (LC–MS–MS)-Based Quantitation of Biopharmaceuticals in Biological Samples
Special Issues
Advances in Liquid Chromatography–Tandem Mass Spectrometry (LC–MS–MS)-Based Quantitation of Biopharmaceuticals in Biological Samples
Liquid chromatography coupled to tandem mass spectrometry (LC–MS–MS) has recently become a more and more popular alternative to traditional ligand-binding assays for the quantitative determination of biopharmaceuticals. LC–MS–MS offers several advantages such as improved accuracy and precision, better selectivity, and generic applicability without the need for raising analyte-directed antibodies. Here we discuss the technical requirements for a successful LC–MS–MS method for the quantitation of biopharmaceuticals and evaluate the advantages and disadvantages compared to ligand-binding assays.


Photo Credit: GI photoStock/GettyImages
The development of proteinâbased pharmaceuticals, or biopharmaceuticals, is by far the fastest growing part of the pharmaceutical industry today. With over 1500 biopharmaceuticals in clinical development and more and more companies shifting their R&D efforts towards this sophisticated and relatively profitable class of drugs, the pharmaceutical landscape has changed beyond recognition compared to 20 or even 10 years ago. As a result, the field of bioanalysis that supports drug development by measuring the concentrations of drugs or relevant endogenous molecules in biological samples has also seen many changes. The quantitative determination of biopharmaceuticals has traditionally been the domain of ligand-binding assays, such as ELISA. However, in the past few years there has been a clear increase in the application of alternative analytical platforms, in particular liquid chromatography coupled to tandem mass spectrometry (LC–MS–MS), which has been the workhorse for small-molecule bioanalysis for over 20 years (1–5). Over the past decade, there have been many advances in the LC–MS–MS-based quantitation of biopharmaceuticals, both from an analytical and a conceptual point of view. In this article, an overview is given of the many aspects of this field of analytical research by reference to a selection of recent applications.
Protein Digestion
Tandem mass spectrometry remains the detection technique of choice for the quantitative determination of biopharmaceuticals because of its sensitivity and its widespread availability in the pharmaceutical and related industries. However, the use of LC–MS–MS to quantify biopharmaceuticals is more complex than for small molecules because it is not directly compatible with molecules with a mass above around 5000 Da. The ions of larger analytes are distributed over many different charge states and usually do not readily fragment and this considerably reduces sensitivity. Therefore, a typical step in the analysis is the (enzymatic) digestion of a biopharmaceutical into a mixture of smaller peptides, followed by the analysis of the digest and quantitation of one or more so-called signature peptides as a measure for the intact protein. Digestion is usually performed using the enzyme trypsin, which cleaves the amino acids chain in proteins after a lysine or arginine. Trypsin is popular because it is readily available at a reasonable price and can cleave proteins into peptides of a size (500–2000 Da) that is well suited for MS–MS detection. Protein digestion enormously increases the complexity of a biological sample. Matrices such as plasma contain proteins at a total concentration of around 80 mg/mL and, when no further clean-up of the sample is performed, each of these proteins is cleaved into a series of peptides that are all of a similar size and have more or less comparable physicochemical and analytical properties. Therefore, it is often challenging to detect low concentrations of a signature peptide in a digest, because of the presence of so many endogenous peptides, which all consist of combinations of the same 20 amino acids and often occur at much higher levels than the signature peptide itself.

85, K.J. Bronsema, R. Bischoff, and N.C. van de Merbel, High-Sensitivity LC–MS/MS Quantification of Peptides and Proteins in Complex Biological Samples: The Impact of Enzymatic Digestion and Internal Standard Selection on Method Performance, 9528–9535 (2013) © American Chemical Society.
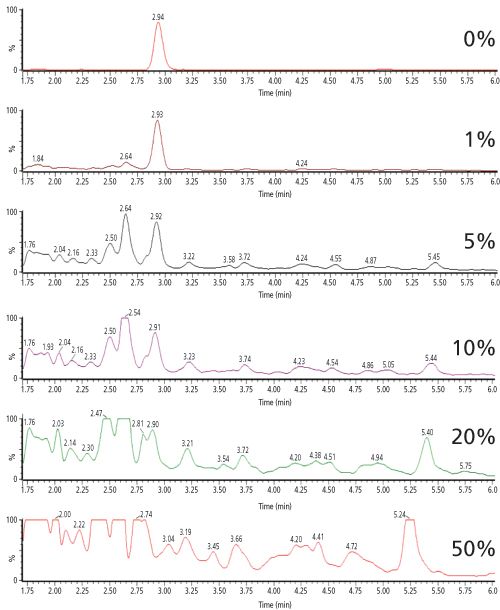
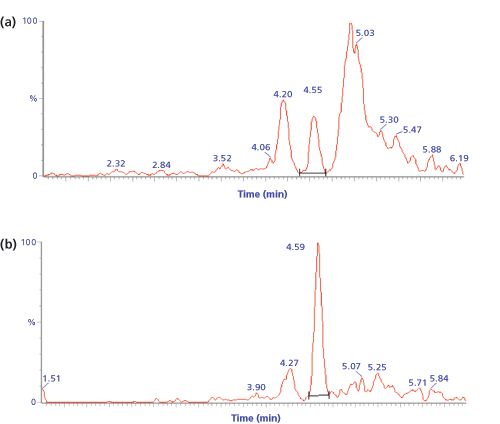
Despite the selective nature of MS–MS detection, chromatograms of digested biological samples often contain many background peaks originating from endogenous peptides that show a response at the mass transition of the signature peptide. Figure 1 shows this effect for a fixed concentration of digested salmon calcitonin in the presence of increasing amounts of digested plasma (6). The selectivity of the method is clearly affected by the presence of endogenous background peptides. As a result, method sensitivity is also heavily impacted - in this case the achievable lower limit of quantitation (LLOQ) increases 100âfold, from 0.2 ng/mL (60 pM) in the absence of matrix peptides to 20 ng/mL (6 nM) in the presence of 50% of digested plasma.

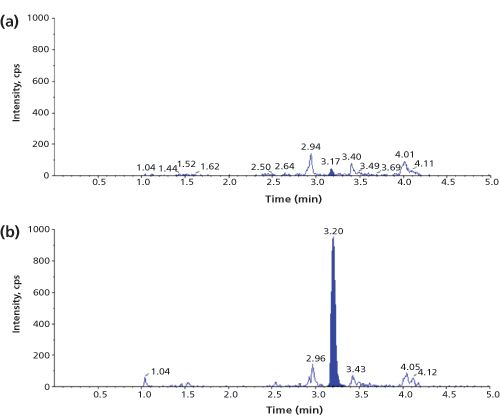
A review of current literature (1,4) shows that a typical LLOQ for a biopharmaceutical in plasma or serum, only treated by digestion, is in the high ng/mL to low µg/mL range (corresponding to low nM levels for many proteins). Figure 2 shows an example chromatogram for a signature peptide of recombinant human alpha-glucosidase at its LLOQ of 0.5 µg/mL (5 nM) in human plasma (7).
Signature Peptide Selection
The possibilities for selecting a proper signature peptide are usually rather limited. First and foremost, it is essential that the selected signature peptide has a unique amino acid sequence that does not naturally occur in any of the endogenous matrix proteins. Selection of a non-unique signature peptide results in an overestimation of analyte concentrations, because the same amino acid sequence that is released from endogenous proteins would contribute to the overall signal. This often disqualifies a large number of the theoretical signature peptides, particularly for biopharmaceuticals with a high degree of similarity to endogenous proteins, such as humanized antibodies (if these need to be quantified in human plasma). In addition, other criteria are applied to ensure robustness of the LC–MS–MS assay. Peptides containing unstable amino acids, such as methionine and tryptophan that can be oxidized, or glutamine and asparagine that can be deamidated, are usually disregarded to avoid losses during analysis, although forced oxidation of a signature peptide to a stable oxidized product has been successfully used (8). Similarly, peptides with (variable) post-translational modifications - such as O- or N-glycosylated amino acids - are typically excluded because these would introduce undesirable heterogeneity. In addition, peptides that are too small, too large, too polar, or too hydrophobic might cause analytical problems because of adsorption, sub-optimal chromatographic behaviour, or limited selectivity and sensitivity. In the end, there may be just a few out of the many potential signature peptides that can be successfully used in practice.
Protein Extraction
An obvious way to improve selectivity and sensitivity of an LC–MS–MS method is to remove interfering matrix proteins prior to digestion, which can be achieved by applying immunocapture (IC) techniques. Magnetic beads or other resins are coated with a protein that displays a high binding affinity towards the analyte, typically an antibody raised against the analyte or the pharmacological target to which a biopharmaceutical is directed. By mixing the sample with a suspension of the beads or passing it through a cartridge filled with the resin, the analyte is selectively isolated from the complex sample. This approach is particularly popular for the quantitation of endogenous proteins such as biomarkers, for which well-characterized immunological reagents are widely available. One example is an LC–MS–MS method for parathyroid hormone (PTH) in human serum (9). A sample of 1 mL was treated by IC with polystyrene beads coated with murine anti-PTH antibodies and the trapped analyte digested with trypsin. The IC treatment allowed the quantitation of PTH down to 40 pg/mL (4 pM) in serum, which shows the enormous clean-up potential of this approach. A completely
15
N-labelled form of PTH was added to the sample as an internal standard at the very beginning of the sample handling procedure. In general, it is desirable that a stableâisotopeâlabelled or other closely related form of the protein analyte be included in the method as an internal standard, to correct for the variability of the extraction procedure. This is also one of the drawbacks of extracting a biological sample before digestion, because such a protein-based internal standard can usually only be obtained by biotechnological means, which may be difficult, if not impossible (4). The disadvantages associated with the use of immunological reagents - such as their potentially limited availability, varying quality, and the interference of matrix proteins with the extraction efficiency - have prompted researchers to investigate alternative so-called antibodyâfree extraction approaches (5). An interesting technique is immobilizedâmetal affinity chromatography (IMAC), which is based on the interaction of metal ions, such as Ni
2+
, with amino acids that feature strong electron donor groups, such as histidine. Proteins with such amino acids on their surface will be selectively captured by IMAC resins. As an example, the biopharmaceutical recombinant human tumour necrosis factor-related apoptosis-inducing ligand (rhTRAIL) has been quantified in human and mouse serum down to 20 ng/mL (340 pM) by removing 95% of matrix proteins, while recovering >70% of the analyte with IMAC (8).

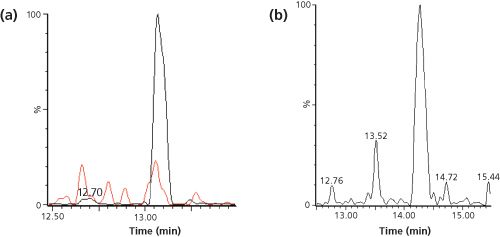
Figure 3: LC–MS–MS (m/z 729.0 to m/z 942.4) chromatograms of the signature peptide of 10 ng/mL rhTRAIL in human serum and the corresponding blanks, pretreated with SCX or IMAC before digestion. Adapted and reproduced with permission from Bioanalysis 7(6), D.Wilffert, R. Bischoff, and N.C. van de Merbel, Antibody-free workflows for protein quantification by LC-MS/MS, 763–779 (2015) © Future Science Ltd.
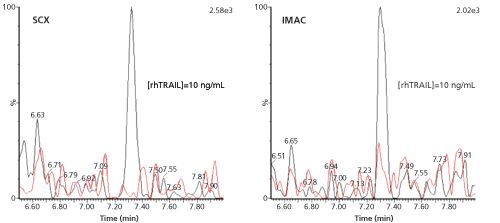
Another technique is solid-phase extraction (SPE) with ion-exchange materials, which separates proteins based on their isoelectric point (pI). Proteins with a relatively high pI bear a net positive charge and can be trapped on a cation-exchange resin at neutral or slightly alkaline pH, at which many endogenous proteins with a lower pI will be negatively charged and thus not be captured. The extraction of rhTRAIL with strong-cation exchange (SCX) SPE was found to have a similar clean-up potential to IMAC, with an analyte recovery of 70% and a protein removal efficiency of 99%. As an illustration, Figure 3 shows chromatograms obtained for 10 ng/mL (170 pM) of rhTRAIL in human serum, which was extracted by SCX or IMAC, followed by trypsin digestion and LC–MS–MS analysis of the signature peptide.
Peptide Extraction
Removal of interfering matrix components is also possible after digestion, that is, at the peptide level. This approach has some distinct advantages. From a practical point of view the optimization of an SPE procedure is more straightforward because of the wide availability of a range of materials that are commonly used for smallâmolecule extractions and because of the more predictable extraction behaviour of smaller peptides compared to that of intact proteins. The accuracy and precision of extractions may be influenced by protein-protein interactions in samples (such as binding of a biopharmaceutical to its target or to anti-drug antibodies), or the occurrence of aggregates. If a sample is first subjected to digestion, these interactions will no longer influence the extraction because all proteins will have been cleaved to peptides that are much less likely to bind to one another with a high affinity. No less importantly, peptide extraction does not need a proteinâbased internal standard; it can instead perform very well when using a stable-isotope labelled form of the signature peptide (4,6), which is considerably less expensive and easier to obtain. It may, however, be difficult to achieve sufficient selectivity because the peptides in a plasma digest are much more similar to each other than the plasma proteins were before digestion. Again, the highest selectivity and sensitivity is achieved by applying immunocapture, which in this case uses immobilized antibodies raised against the signature peptide. This approach is most widespread in the field of biomarker analysis, where the number of analytes is relatively limited and assays are relevant to many research groups around the globe. Large clean-up efficiencies can be achieved in this way, as was reported for the endogenous proteins α1-antichymotrypsin (1453âfold enrichment relative to matrix proteins) and TNF-α (573-fold enrichment) (10).
IC at the peptide level is less popular in biopharmaceutical analysis, probably because of the general drawbacks of antibodyâbased reagents with regard to availability and batch-to-batch reproducibility. A more generic approach for peptide extraction from a digest is to use conventional ion-exchange SPE, but this needs to be carefully optimized to obtain sufficient selectivity. A digest of a protein-rich biological sample (such as plasma) contains a multitude of peptides, which all have carboxylic and amine groups, and the signature peptide can only be separated from the excess of endogenous background peptides if its pI value is sufficiently different. Typically, the pH and ionic strength of the loading, washing, and elution steps need to be carefully optimized for a selective extraction.

7(1), K.J. Bronsema, R. Bischoff, M.P. Bouche, K. Mortier, and N.C. van de Merbel, High-sensitivity quantitation of a Nanobody® in plasma by single-cartridge multidimensional SPE and ultra-performance LC-MS/MS, 53–64 (2015) © Future Science Ltd.
A biopharmaceutical nanobody was quantified down to 10 ng/mL (360 pM) in rabbit and human plasma by trypsin digestion followed by SPE on a weak-anion exchange phase (11). The signature peptide contained three carboxylic acid groups and was strongly retained by the positively charged SPE phase at pH 5; many endogenous peptides with less negative charges were not trapped during sample loading or were removed from the SPE material by a washing step with 300 mM sodium chloride. The mixed-mode SPE phase, which also contained reversed-phase groups, was subsequently neutralized at a high pH and the (relatively polar) signature peptide was eluted, while some less polar endogenous peptides remained bound by reversedâphase interactions. In this way, two dimensions of selectivity (ion exchange and reversed phase) were used to isolate the signature peptide from the plasma digest. Figure 4 illustrates that many interfering peaks were removed from the chromatogram with this approach and that selectivity was clearly improved. Of course, cation-exchange SPE can be applied in the same way in case the signature peptide has multiple positive charges, and even reversedâphase SPE might be an option if the signature peptide is particularly hydrophobic.
Combined Protein and Peptide Extraction

As illustrated above, generic protein or peptide extractions typically result in LLOQs in the low ng/mL (mid to high pM) range, while IC extraction at the protein or peptide level enables quantitation down to mid pg/mL (low to mid pM) concentrations. If more sensitivity is required, one option is to combine protein and peptide extractions. Excellent selectivity and sensitivity can be reached even without antibody-based extraction materials, as was shown for rhTRAIL in saliva (12). After IMAC extraction of the protein analyte and trypsin digestion, the digest was further purified using SPE on a SCX cartridge. Because of the presence of four basic amino acids in the signature peptide, the digest was acidified before loading onto the SPE phase. The peptide was then trapped and endogenous peptides were removed by washing with 200 mM sodium chloride. After elution at alkaline pH, the signature peptide was quantified using LC–MS–MS. As shown in Figure 5, a TRAIL concentration as low as 0.2 ng/mL (3.4 pM) could be quantified in both dog and human saliva. In principle, protein or peptide extractions can be combined in many ways and as long as the separation mechanisms are orthogonal, improved selectivity and sensitivity can be expected compared to a single-extraction approach.

Figure 6: LC–MS–MS chromatograms of two mass transitions (m/z 592.8 to m/z 943.5 in red and m/z 592.8 to m/z 830.5 in blue) of the signature peptide of IL-21 in human serum; (a) blank and (b) 0.78 pg/mL. Samples pretreated with immunocapture both before and after digestion. Adapted and reproduced with permission from Analytical Chemistry 85, J. Palandra, A. Finelli, M. Zhu, et al., Highly Specific and Sensitive Measurements of Human and Monkey Interleukin 21 Using Sequential Protein and Tryptic Peptide Immunoaffinity LC–MS/MS, 5522–5529 (2013) © American Chemical Society.
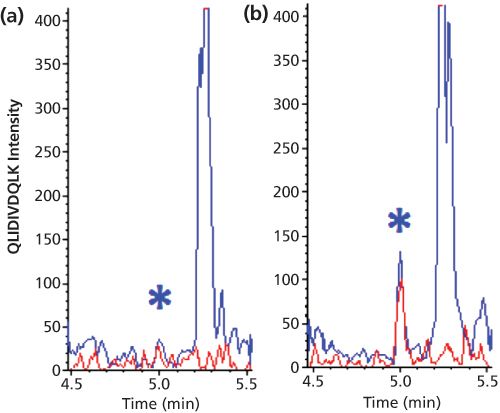
The ultimate combination of protein and peptide extraction is IC of the protein analyte followed by digestion and IC of the signature peptide. Although this requires two specifically raised antibodies and is by no means a generic approach, it can result in impressive sensitivities. The biomarker interleukin-21 (ILâ21) was quantified in human serum and monkey tissues with an LLOQ of 0.78 pg/mL (0.05 pM). This was achieved by combining offâline magnetic bead-based protein extraction using an anti-IL-21 antibody with on-line enrichment of the signature peptides using immobilized anti-peptide antibodies (13). Figure 6 shows representative chromatograms. It is important to realize that the obtained LLOQ corresponds to a molar concentration of the protein, which is five orders of magnitude lower than that shown in Figure 2 (digestion only). This convincingly demonstrates the enormous clean-up capability of this combination of techniques.
LC–MS–MS versus ELISA
Compared to ligand-binding assays, LC–MS–MS has a number of analytical advantages such as a larger linear dynamic range; (usually) higher accuracy and precision because of the possibility to apply internal standards (4); the ability to quantify multiple analytes simultaneously; and the fact that it does not necessarily require immunological reagents (5). The last point can be especially critical, because such reagents may be problematic to obtain or show a large batch-to-batch variability, which makes comparison of results between laboratories or over a longer period of time difficult, if not impossible. The disadvantages of LC–MS–MS include its generally higher operational cost; more limited sample throughput; and less favourable concentration sensitivity. In addition, with the digestion step that is generally needed for LC–MS–MS, the threeâdimensional structure of a protein analyte is lost and the analytical principle is therefore not related to the complex molecular structure of a protein, which determines its pharmacological activity. Now that more and more reports are appearing that compare newly developed LC–MS–MS methods with existing ELISAs for the same protein analyte, it is becoming increasingly clear that both techniques do not always give superimposable concentration results (14,15). Although in the world of small-molecule quantitation, two different results for the same sample would be seen as proof that at least one of them is incorrect, this is not necessarily true for biopharmaceuticals. It should be realized that, in contrast to small molecules, LC–MS–MS as well as ELISA only use a small part of the protein molecule for the actual quantitation, the signature peptide and the binding epitope, respectively, and this may represent as little as a few percent of the entire molecule. Furthermore, both techniques are based on quite different (bio)chemical principles, to which the structurally complex and often heterogeneous biopharmaceuticals may respond in different ways. Thus, neither LC–MS–MS nor ELISA should be regarded as the ultimate quantitation technique for biopharmaceuticals, but rather as complementary tools for obtaining quantitative information about this complicated but very interesting class of compounds.
Acknowledgement
Stichting Technische Wetenschappen (STW) and Samenwerkingsverband Noord-Nederland (SNN) are gratefully acknowledged for providing financial support for part of the work described in this paper.
References
(1) R. Bischoff, K.J. Bronsema, and N.C. van de Merbel,
Trends Anal. Chem.
48
, 41–51 (2013). (2) G. Hopfgartner, A. Lesur, and E. Varesio,
Trends Anal. Chem.
48
, 52–61 (2013). (3) I. van den Broek, W.M. Niessen, and W.D. van Dongen,
J. Chromatogr. B
929
, 161–179 (2013). (4) K.J. Bronsema, R. Bischoff, and N.C. van de Merbel,
J. Chromatogr. B
893–894
, 1–14 (2012). (5) D. Wilffert, R. Bischoff, and N.C. van de Merbel,
Bioanalysis
7
, 763–779 (2015). (6) K.J. Bronsema, R. Bischoff, and N.C. van de Merbel,
Anal. Chem.
85
, 9528–9535 (2013). (7) K.J. Bronsema, R. Bischoff, W.W.M.P. Pijnappel, A.T. van der Ploeg, and N.C. van de Merbel,
Anal. Chem.
87
, 4394–4401 (2015). (8) D. Wilffert, C.R. Reis, J. Hermans, N. Govorukhina, T. Tomar, S. de Jong, W.J. Quax, N.C. van de Merbel, and R. Bischoff,
Anal. Chem.
85
, 10754–10760 (2013). (9) V. Kumar, D.R. Barnidge, L.S. Chen, J.M. Twentyman, K.W. Cradic, S.K. Grebe, and R.J. Singh,
Clin. Chem.
56
, 306–313 (2010). (10) J.R. Whiteaker, L. Zhao, H.Y. Zhang, L.C. Feng, B.D. Piening, L. Anderson, and A.G. Paulovich,
Anal. Biochem.
362
, 44–54 (2007). (11) K.J. Bronsema, R. Bischoff, M.P. Bouche, K. Mortier, and N.C. van de Merbel,
Bioanalysis
7
, 53–64 (2015). (12) D. Wilffert, unpublished results (13) J. Palandra, A. Finelli, M. Zhu, J. Masferrer, and H. Neubert,
Anal. Chem.
85
, 5522–5529 (2013). (14) N.C. van de Merbel, K.J. Bronsema, and M. Nemansky,
Bioanalysis
4
, 2113–2116 (2012). (15) P. Bults, N.C. van de Merbel, and R. Bischoff,
Expert Rev. Proteomics
12
, 355–374 (2015).
Nico van de Merbel
is scientific director at the bioanalytical laboratories of PRA Health Sciences (Assen, The Netherlands and Lenexa, KS, USA) and honorary professor at the University of the Groningen, The Netherlands.














Silvia Radenkovic on Building Connections in the Scientific Community
April 11th 2025In the second part of our conversation with Silvia Radenkovic, she shares insights into her involvement in scientific organizations and offers advice for young scientists looking to engage more in scientific organizations.



Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

