A Review of MOSH and MOAH Analysis in Food
An overview of the analytical approaches proposed, from sample preparation to the final chromatographic determination, for the reliable risk assessment of mineral oil hydrocarbon (MOH) contaminants in food. The analysis of these contaminants in food is a challenging task and requires a comprehensive approach to tackle the cumbersome issues related to their determination.
Mineral oil hydrocarbons (MOH) are a very complex mixture of isomers classified into mineral oil saturated hydrocarbons (MOSH) and mineral oil aromatic hydrocarbons (MOAH). The analysis of such contaminants in food is a challenging task and requires a comprehensive approach to tackle the cumbersome issues related to their determination. Additionally, their toxicity is still under investigation and requires further studies supported by more detailed analytical data. This review aims to give an overview of the different analytical approaches proposed, from sample preparation to the final chromatographic determination, to respond to the request of consumers and institutions, such as the European Food Safety Authority and European Commission, for a reliable risk assessment. Emphasis is given to hyphenated chromatographic techniques as powerful tools to gain deeper insights into the MOSH and MOAH problem.
The International Agency for Research on Cancer (IARC) defined mineral oils (MO) as follows: “Mineral oils, which are also known as base oils, mineral base oils or lubricant base oils, are chemical substances prepared from naturally occurring crude petroleum oil. Crude oil is distilled first at atmospheric pressure and then under high vacuum to yield vacuum distillates and residual fractions that can be further refined to mineral oils” (1). The strict definition of contamination by mineral oil hydrocarbons (MOH) refers to the accidental presence in food of crude petroleum and other products derived from it by distillation and refining process, for example, diesel fuel, jet fuel, white oils, lubricants, and solvents. Despite that, the quantification of MOH can, sometimes, erroneously include analogue mixtures—the use of which is permitted in food contact material (FCM) and in the food industry—such as polyolefins and white oils (highly refined mineral oil where the presence of aromatic compounds is minimized). Therefore, a careful interpretation of the results is needed.
From an analytical viewpoint MOH are divided into two main fractions, namely MOSH (MO saturated hydrocarbons, composed by linear, branched, and alkyl-substituted cycloalkanes) and MOAH (MO aromatic hydrocarbons, which include mainly alkyl-substituted [poly] aromatic hydrocarbons with a different number of fused rings).
Food is the main source of MOH intake (2) and various studies were conducted to evaluate the exposure to mineral oils over time, both on animal tissues (3–9) and human tissues (10–13), highlighting accumulation of MOSH in the liver, spleen, lymph nodes, and adipose tissue. The end point was identified as hepatic micro-granulomas based on studies on Fischer 344 rats, but later it was considered irrelevant to humans due to inter-species differences in terms of MOSH absorption, catabolism, and sensitivity (12). MOAH are classified as potentially mutagenic, referring in particular to 3-7 ring compounds (2). A high degree of alkylation is thought to reduce their metabolic activation, leading to the formation of non-mutagenic intermediates (13).
In 2012 the European Food Safety Authority (EFSA) published its first opinion about MOH in food (2) but no definitive conclusions were drawn because of the limited information available.
In 2017, the EFSA and the European Union (EU) (14) required the collection of more data to characterize the sub-classes present in the MOH fractions and so evaluate better the risk, but guidance for harmonized data collection and report were only published in February 2019 (15). In this guidance, the carbon range of MOH was analyzed. This guidance recommended to quantify the C10–C50 carbon range. In 2019, the EFSA released a new opinion, following the Foodwatch report on the presence of MOAH in infant and follow-on formulas (16), still concluding that characterization of hazards is not possible in the absence of relevant dose-response data and information regarding the presence of the more health-concerning 3-7 ring MOAH (17).
The situation is also complicated by the permitted use of white mineral oils as food additives (microcrystalline wax and hydrogenated poly-1-decene) (18), food processing aids (mold releasing and anti-caking agents), and FCM additives
(19). Moreover, some paraffin oils and mineral oils are allowed to be used as pesticides in the production of organic food (20,21).
While a common regulation about migration in food does not exist yet, some national law can be considered. Specific limits were set for the migration of MOH from inks, by the Swiss Printing Ink Ordinance (RS 817.023.21), and from FCM, by the 22nd German Federal Ministry of Food and Agriculture (BMEL) ordinance. In Belgium,
the Scientific Committee of the Federal Agency for the Safety of the Food Chain (FASFC), published “Advice 19-2017”, which sets action thresholds between 5–150 mg/kg for the MOSH fraction (C16-C35), depending on the food category (22).
Except for the method EN 16995 (23) for MOSH and MOAH determination in vegetable oil, no official methods exist.
In this context, the goal of this review is to provide a current overview on the topic, with particular focus on the analytical perspective. Emphasis will be given to the most innovative and recent solutions proposed to tackle this issue. Regarding the sample enrichment/purification and MOSH/MOAH separation, a premise has to be made since they are often achieved simultaneously by a single step. For the purposes of logic, they will be separated in two different sections, and discussed as individual treatments. Moreover, a further subdivision in offline and online methods will be done.
Extraction of Mineral Oil Hydrocarbons
Since MOH are ubiquitous contaminants, the possible contribution of additional contamination should be minimized through a conscious choice of solvents (high purity grade), a proper cleaning of the glassware, and high care and possible minimization of the sample handling.
In the extraction step, hexane is by far the most commonly used solvent. It can be utilized alone, in sequence, or mixed with other solvents (such as ethanol), depending on the fat content, moisture of the matrix, and on the type of contamination investigated (superficial migration from FCM or inner contamination). It is used in MOH extraction from many matrices, including cereal or cereal-based dry products, wet foods, oils, fats, and FCM (24,25).
In the case of dry foods, superficial contamination can be easily extracted by direct immersion in hexane, whereas for the inner contamination, prior rehydration of the matrix is required (for example, overnight at 80 °C). Then, for wet or rehydrated matrices, a conditioning step in ethanol (1 h) is fundamental to replace the immobilized water and thus mediate the hexane entrance to the pores. Another positive effect of the ethanol is its swelling ability towards starch and denaturing power towards proteins, thus favoring the release of entrapped hydrocarbons (26,27).
For edible oils and fats, a simple dilution in hexane is performed, followed by online or offline purification steps. Sample enrichment is often performed to increase sensitivity, and auxiliary purification steps can be required to remove interference such as natural alkanes and olefins.
Another scenario is represented by the extraction of MOH from FCM. Recycled papers and paperboards are generally left to soak in a solution of hexane/ethanol (1:1 v/v) (28,29). The conditions are opportunely chosen to reduce the extraction of high molecular weight hydrocarbons that could remain in the retention gap, causing a carryover effect and appearing randomly as broad unresolved peaks (30). In the case of plastic films, the extraction time with pure hexane must be reduced to the minimum, according to film thickness and permeability, in order not to extract excessive amounts of plastic oligomers.
Rapid, alternative, and efficient MOSH and MOAH extraction can be obtained by microwave assisted saponification (MAS) or pressurized liquid extraction (PLE). MAS with simultaneous solvent extraction was proposed instead of the classical saponification followed by solvent partitioning (31). Different cereal-based foodstuffs were saponified with methanolic potassium hydroxide (KOH) (120 °C × 20 min) in the presence of hexane. The hexane supernatant was directly collected for analysis.
PLE has been proposed as an alternative in dry foods with low fat content (27). The external contamination, deriving from packaging migration, was differentiated from the internal one. Hexane was used to recover the external contamination, while the total contamination was determined by a two-cycle PLE in hexane/ethanol (1:1 v/v) at 100 °C for 5 min, obtaining comparable results to the overnight extraction previously proposed (26). In the case of cardboard (32), MOH were extracted at 60 °C for 5 min, by a two-cycle PLE in hexane, reducing the extraction time compared to the previous method (28).
Sample Enrichment and Purification
Often additional purification steps are required, but they may lead to cross-contamination, thus reducing the accuracy. They should be appropriately applied and only when strictly necessary. Support in this regard is given by the “decision tree” reported in the JRC Guidance (15). The enrichment and auxiliary techniques are herein briefly summarized.
Offline Enrichment and Purification
Enrichment: It is required to increase the sensitivity by removing the bulk of the triglycerides. Saponification followed by partitioning in hexane is usually applied (31). Alternatively, offline solid-phase extraction (SPE) with activated silica has been proposed (33,34). Performance of the activated silica is higher compared to the non-activated one and comparable to that of silver silica in terms of fat retaining capacity, the latter also improving olefins retention, thus reducing the need for an additional epoxidation step (33).
Natural alkanes removal: Naturally-occurring alkanes, generally ranging from C21 to
C33 (with odd carbon numbers prevailing over the even ones), are found mostly in vegetable matrices. They are usually subtracted a posteriori from the integration of the MOSH hump. However, activated aluminum oxide must be used to remove n-alkanes greater than C20, without affecting the iso-alkanes fraction, when they overload the chromatogram (35).
Olefins removal: This includes highly unsaturated natural olefins, for example, squalene, carotene, and sterenes, which interfere with MOAH, and unsaturated molecules released by polyolefin packaging, such as polyolefin mono-unsaturated hydrocarbons (POMH) and poly alpha olefins (PAO), coeluting with the MOSH fraction. It has to be specified that polyolefin packaging also releases polyolefin saturated hydrocarbons (POSH).
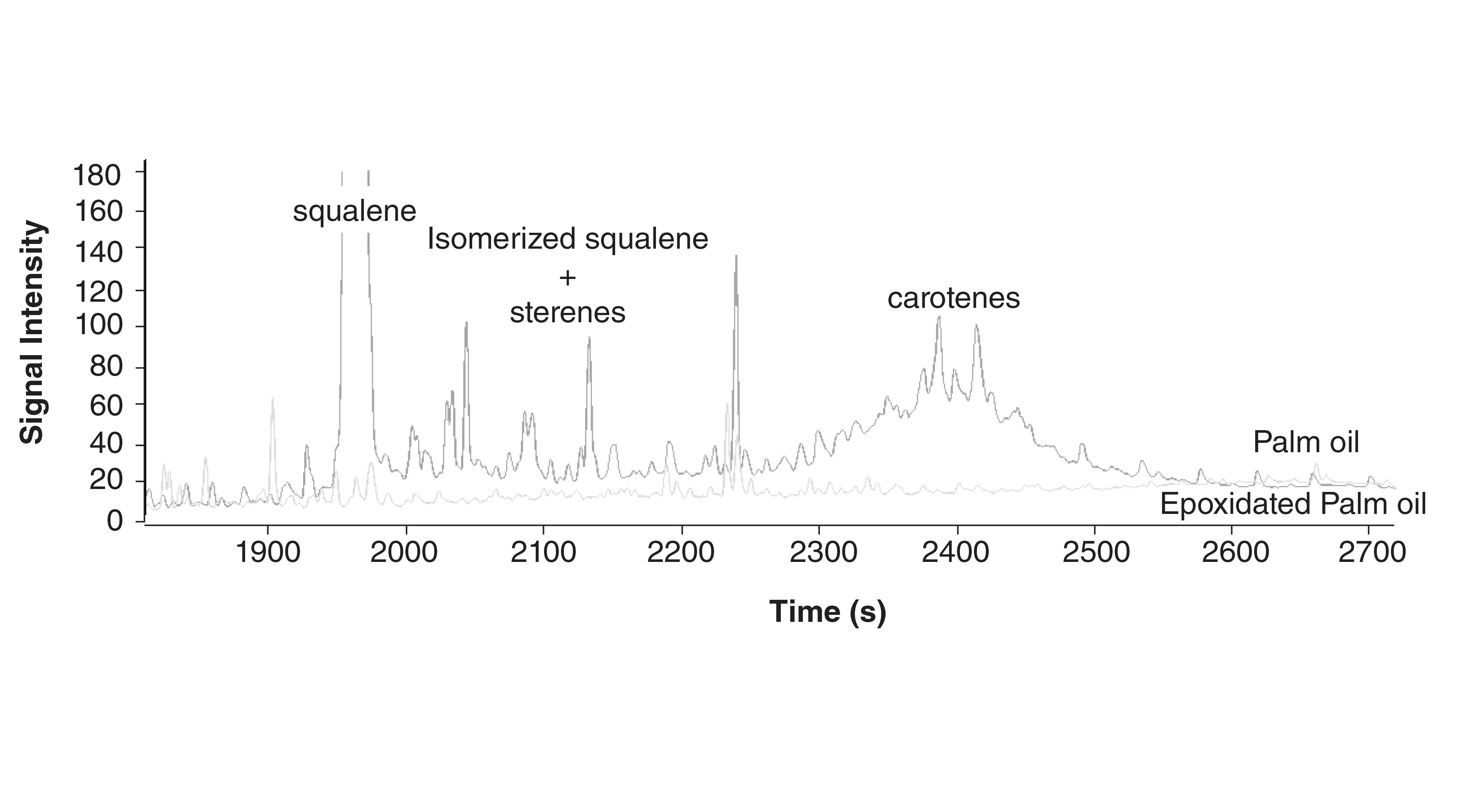
The removal of interfering olefins was initially performed, increasing their polarity by bromination of the double bonds. This reaction also significantly affected the MOAH fraction, causing loss of analytes (36), so epoxidation with meta-chloroperbenzoic acid (mCPBA) was proposed to assure a better selectivity toward aliphatic double bonds, yet a possible loss of 20–35% of MOAH may occur during the reaction (37–39). Figure 1 shows the profile of a palm oil sample before and after epoxidation.

 and after (green trace) epoxidation.")
FIGURE 1: GC-FID profile of the MOAH fraction of a palm oil sample before (orange trace) and after (green trace) epoxidation.
Online Enrichment and Purification
Enrichment: Applying the routine liquid chromatography–gas chromatography (LC–GC) method (described in detail later), which exploits the fat retention of an LC silica column (25 cm × 2.1 mm, 5-μm), food extracts can be directly injected into the LC column as long as the fat content is lower than the column capacity (~20 mg) and the limit of quantification (LOQ) for MOH is reached (~50–100 ng on column with the flame ionization detector) (30,37,40). When this condition is not fulfilled, enrichment procedures are necessary. The use of a large LC silica column (25 cm × 4.6 mm, 5-μm) to increase the fat retaining capacity (from ~20 to 150–200 mg) was proposed (41), but the eluted hydrocarbon fraction had a volume of 6 mL, which is difficult to handle in the online coupling with gas chromatography (GC). Thus, offline methods are usually, preferred for enrichment purposes.
Natural alkanes removal: Fiselier et al. proposed the use of an online alumina oxide LC column for removal of n-alkanes from the MOSH fraction (42). In this case, a primary LC silica column (25 cm × 2 mm, 5-μm) was designed to retain fats and pre-separate MOSH from MOAH. Only the saturated fraction was sent to the secondary aluminum oxide column (10 cm × 2 mm, 63–200-μm) activated at ~400 °C, where the long-chain n-alkanes were retained. Compared to the offline method, a great advantage is the possibility to restore the alumina column by a backflush step in iso-octane, allowing operation for many cycles and, of course, reducing the potential contamination.
Olefins removal: An LC–LC–GC method (43) for the analysis of POMH in food and FCM was proposed. After the separation of MOSH and MOAH in a silica column, the MOSH fraction was sent to a secondary silver silica column, that allowed the isolation of the POMH. The MOAH fraction instead, bypassed the silver silica path being directly sent to the GC in order to avoid the possible coelution of the POMH with the following MOAH fraction.
A similar approach was exploited for the removal of natural olefins interfering with the MOAH fraction. In this case, the MOAH fraction was diverted to a secondary silver silica column to remove olefin, while the MOSH fraction was directly sent to the GC–flame ionization detection (GC–FID) system for quantification (44).
Conventional MOSH and MOAH Separation
Offline SPE: Offline SPE methods followed by injection in GC have been developed to meet the instrumental availability of many laboratories that may not afford an online system.
In 2011 Moret et al. (33) published an offline SPE method for MOSH determination in vegetable oils, using a laboratory-made glass cartridge filled with 1 g of silver silica sorbents. The method was later extended to the separation of MOSH and MOAH, eluting the MOSH fraction (1.5 mL) with hexane, and the MOAH fraction (7 mL)
with hexane/dichloromethane 1:1 (45). However, for some samples, the method might fail in retaining esterified fatty acid or waxes, limiting its applicability (46).
Fiselier and co-workers (34) overcame the problem by using 3 g of 0.3% silver silica, allowing the analysis of samples containing up to 20% fat, using a mixture of toluene:dichloromethane:hexane (0.5:2:7.5 v/v) as eluent.
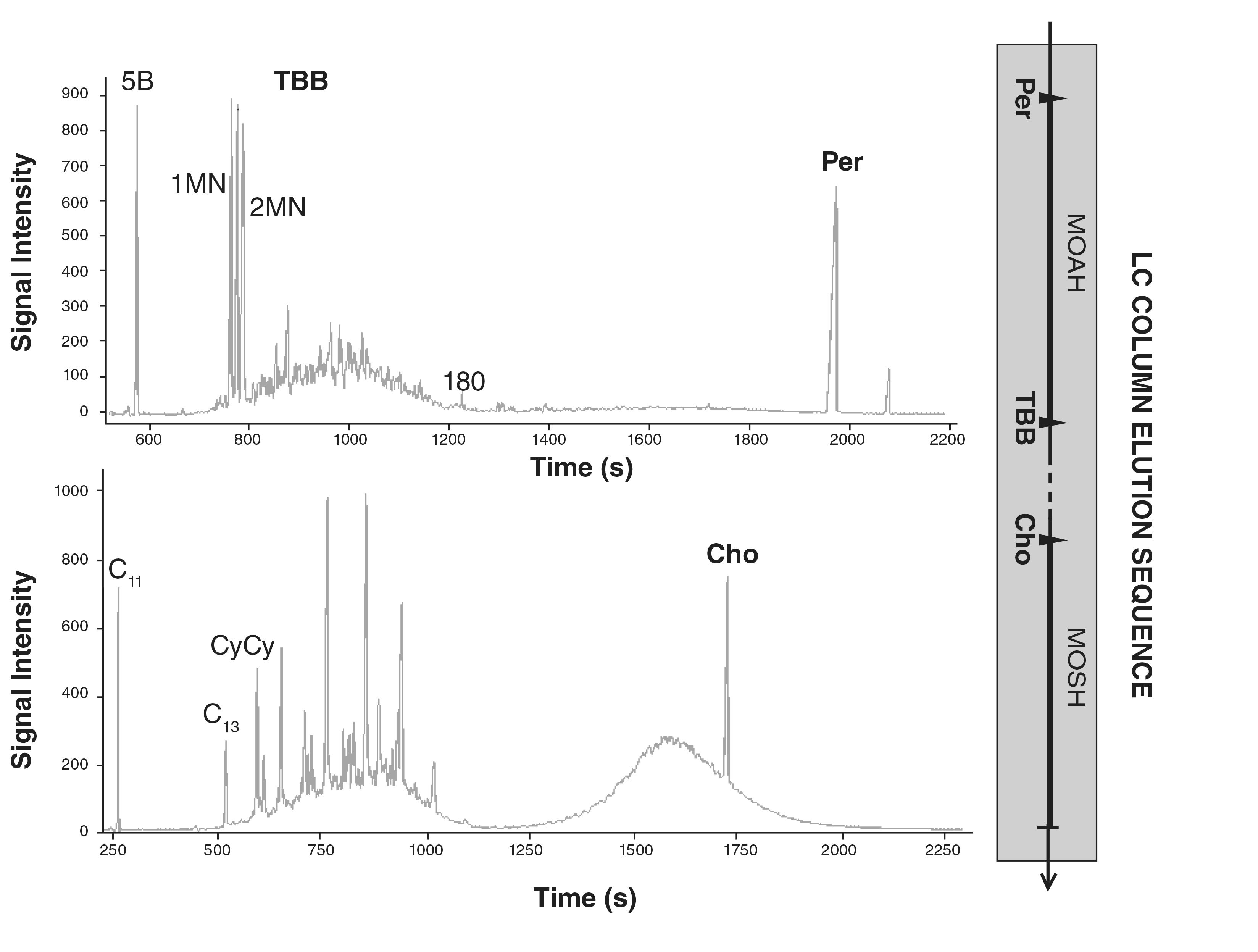
Online LC–GC: Online LC to isolate MOSH and MOAH fractions, followed by GC–FID, is the method of choice for quantification (30,37,40). MOSH are the eluted first, followed by the MOAH, while triglycerides and more polar compounds are retained and backflushed with dichloromethane. Internal Standards (ISs) are used
to identify the boundaries of the MOSH and MOAH fractions from the LC (Figure 2): cholestane (Cho) is used to mark the end of the MOSH fraction. Tertbutylbenzene (TBB) and perylene (Per) are used as markers of the beginning and the end of the MOAH fraction, respectively (37). Additional ISs are used for quantification purposes and quality control of the entire LC–GC transfer process, based on the assessment that specific quantity ratios are verified at each analysis. Undecane (C11) and bicyclohexyl (CyCy) are used for quantification of the MOSH hump while 1-methylnaphthalene (1MN) and 2-methylnaphthalene (2MN) for the MOAH. C11 and 5B (n-pentylbenzene) are used for the MOSH and MOAH fraction, respectively, as watchdogs for volatile losses. Recently it has been reported that CyCy is a more suitable marker for the end of the LC MOSH fraction than Cho, as it elutes slightly later (40). DEHB (1,4-di(2-ethylhexyl)benzene) substituted the use of TBB in order to include the small amount of highly alkylated MOAH that elute right after the latest MOSH fractions.

FIGURE 2: Representation of the MOSH and MOAH fractions eluting from the LC silica column, and their corresponding GC–FID analysis. Cholestane (Cho) marks the end of the MOSH fraction. Tertbutylbenzene (TBB) and perylene (Per) mark the beginning and the end of the MOAH fraction, respectively. CyCy is used for MOSH quantification. 1MN and 2MN are used for MOAH quantification.
The critical step in the hyphenation of LC with GC is the transfer of a large volume of solvent eluted from the LC into the GC column. “On-column” and “Y” interfaces emerge among the other interfaces since they allow the coverage of a broader range of volatility without discrimination or losses of volatiles (30,47).
The GC separation column is preferably not polar (for example, 100% PDMS or 95% methyl, 5% phenyl), ranging from 10 to 30 m in length with thin-film thickness (for example, 0.15 μm) to reduce column bleed and facilitate the elution of high boiling components.
Since both MOSH and MOAH form a hump of unresolved peaks, a fast temperature gradient (~20–40 °C/min) is set to obtain the maximum sensitivity and avoid hump broadening. A high gas pressure helps to elute high boiling hydrocarbons.
The FID detector is used for quantification purposes because it gives virtually the
same response factor for all the hydrocarbons of interest, rendering the calibration simplified by the use of appropriate standards.
On the other side, mass spectrometry (MS) is required for confirmatory purposes (in accordance with EU Recommendation 657/2002) (48), however similar compounds
can present very different response factors (for example, hexane and cyclohexane, or MOAH with different alkylation, such as dimethyl or ethyl-), thus limiting the use of MS for quantification of the complex mixture deriving from the MOSH and MOAH contamination. Nevertheless, MS it is used to confirm the presence of specific markers such as pristane, phytane, diisopropyl naphthalene (DIPN), dibenzothyophene (DBT), and hopanes.
Comprehensive 2-D GC based methods
In 2009, Biedermann and Grob introduced GC×GC in the field of MOH analysis to investigate in detail the MOAH fraction of a highly contaminated Ukrainian sunflower oil (49). Offline GC×GC after HPLC pre-separation allowed classifying the unresolved mixture according to the number of aromatic rings and the degree of alkylation with an apolar column in the first dimension (1% vinyl, 99% dimethyl polysiloxane) and a mid-polar column in the second dimension (50% dimethl polysiloxane, 50% biphenyl).
GC×GC provides an enhanced resolution, allowing the separation of all the sub-classes within the MOSH and the MOAH fraction. However, the two fractions need
to be analyzed separately due to the different concentration factor (usually in the 4:1 ratio) and to avoid the coelution of four- and five-ring saturated hydrocarbons, such as steranes, hopanes, and bicyclic sesquiterpenes with the highly alkylated two- and three-ring aromatics (50). Differently from the first application, the preferred GC×GC columns combination is medium polar × apolar to maximize the resolution in the MOSH fraction and easily differentiate between MOSH and synthetic hydrocarbons (POSH and PAO) (50).
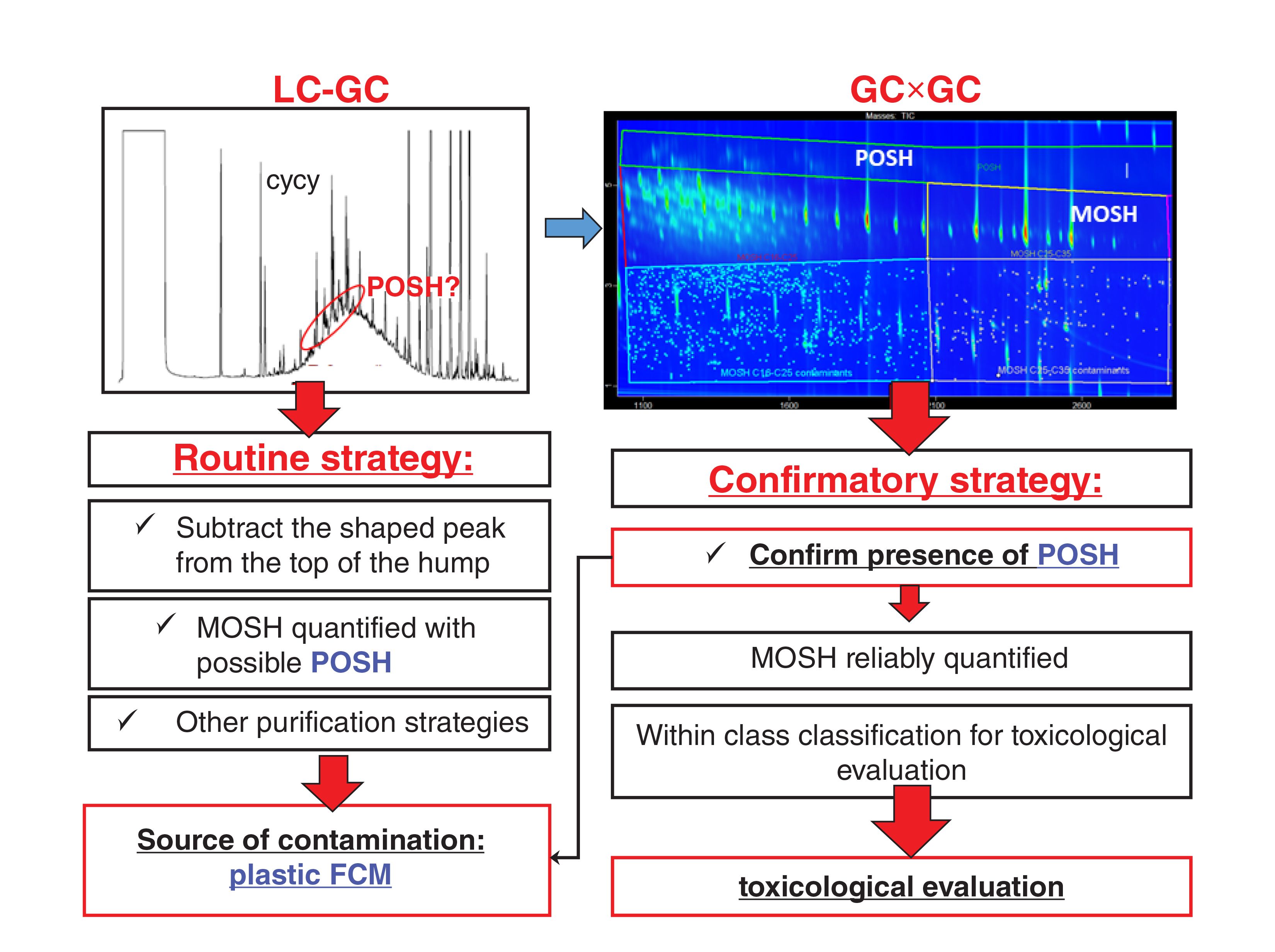
A GC×GC system, coupled with simultaneous dual detection— namely MS and FID—has recently been proposed (51). The two 2-D plots obtained were examined in combination to provide complementary and confirmatory information. A flowchart of the procedure is shown in Figure 3 for the MOSH fraction. The 2-D characteristic profile of both the MOSH and the MOAH fraction supports the determination of the possible sources of contamination in food, providing indications about the kind of MOH present and the type of refining they underwent (50), as well as giving additional information on the sub-classes of the MOSH and MOAH fractions for toxicological purposes. In the specific example of Figure 3, the additional class separation obtained in the 2-D space allowed easy separation of POSH from the MOSH and provided information on the distribution between n-, iso-, and cyclic alkanes.

FIGURE 3: Comparison of the interpretation process of the LC–GC and GC×GC–FID/MS traces of the MOSH fraction of a spice sample. In the 2D plot the POSH are eluted above the MOSH fraction and can be easily removed from the quantification of MOSH.
For volatile contamination coming from cardboard, quantification
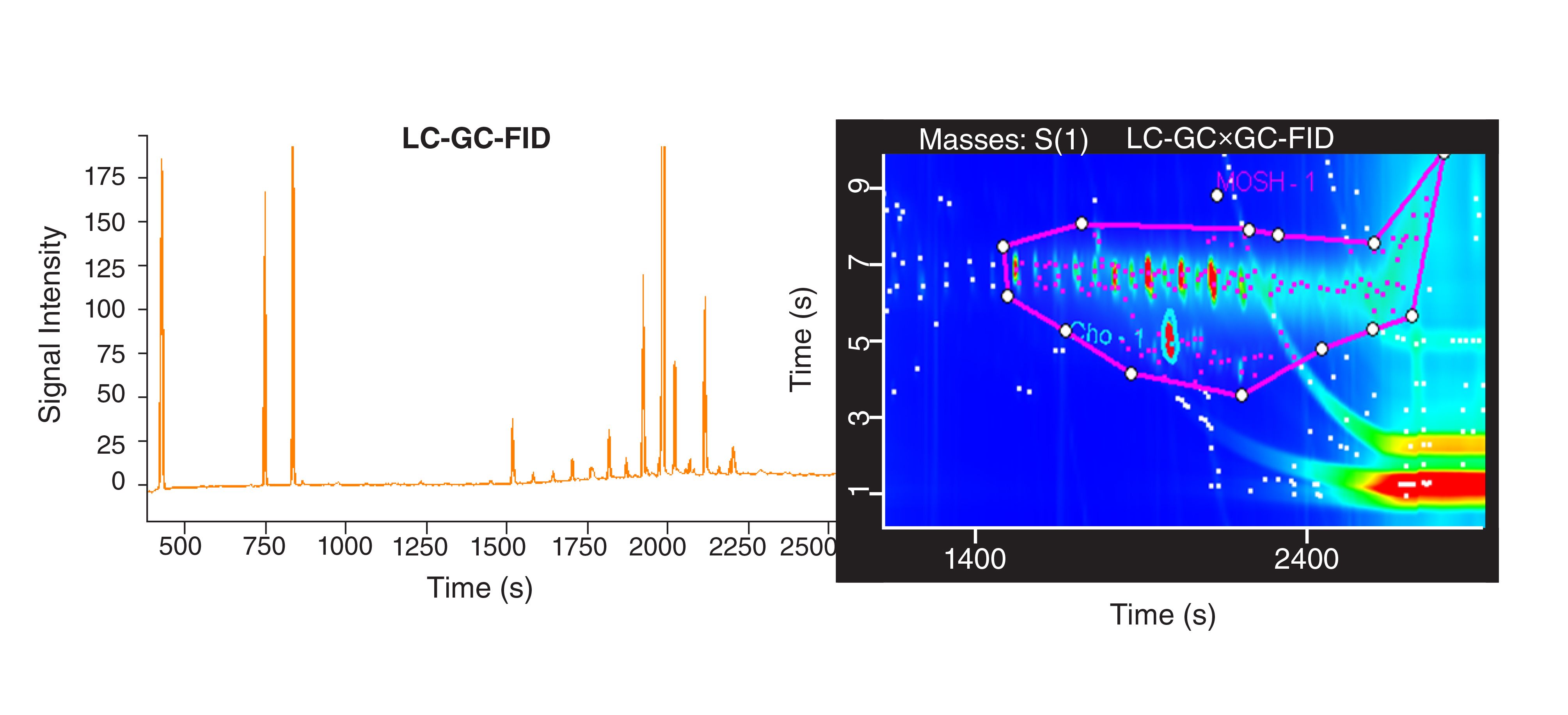
in GC×GC–FID was proposed, obtaining results comparable to the LC–GC–FID (46). More recently, a comprehensive platform—namely LC–GC×GC–TOFMS/FID—has been developed to face the analytical challenge of MOSH and MOAH fractionation, characterization, and quantification in a single analysis (51). Works towards the validation of the quantitative approach are ongoing with promising preliminary results. Figure 4 shows the comparison of an LC–GC–FID trace and an LC–GC×GC–FID trace obtained from the same instrument: quantification of the MOSH hump, subtracted by the alkanes on top of the hump, provided comparable results, namely 9.6 and 9.1 μg/g, respectively.

FIGURE 4: Comparison of LC–GC–FID and LC–GC×GC–FID trace chromatograms of the MOSH fraction of a palm oil obtained using the same LC-GC×GC platform in the 1D and 2D mode.
Alternative Techniques
As previously mentioned, completely automated LC–LC–GC methods using a secondary silver-silica column were proposed to efficiently remove interferents from the MOSH fraction (such as POMH) (43) and the MOAH fraction (such as olefin) (44) for the analysis of food. Recently, the retention mechanism of the silver silica column was investigated as a first dimension of a comprehensive LC×GC system, alternatively coupled to a flame ionization detector or vacuum ultraviolet (VUV) detector. Small fractions (167 μL) of the eluate from the LC were collected and, offline, transferred to the GC system (1 μL injection in splitless mode). The 2-D plot was generated using a programming platform for computational mathematics (52). The silver silica column allowed a group-type separation based on the degree of aromaticity; moreover, the use of the VUV added an extra level of information to the FID.
Later, the same silver silica stationary phase was employed to separate MOSH and MOAH using a supercritical fluid chromatographic (SFC) system coupled with FID and UV (53). Despite a less efficient separation of the MOSH and MOAH fractions, the use of the dual FID/UV detection allowed a deconvolution procedure by subtracting the UV signal from the FID signal, to eliminate the contribution of the aromatic fraction coeluted in the MOSH fraction. Both methods were explored in pure MOH for food and cosmetic applications.
Of high importance, in accordance with the request of the EFSA and the EU, is the possibility to reliably quantify MOAH in sub-classes, with particular emphasis on the 3-7 rings family. Moret et al. proposed an online LC–LC–GC method (41). A first large silica column (250 × 4.6 mm i.d.) retained triglycerides eluting MOH in 6 mL of pentane. An online solvent evaporator (SE) packed with silica gel guaranteed the evaporation of the solvent without loss of the most volatile components. The concentrate fraction was then transferred to an amino LC column, for further separation according to the ring number, and then online transferred to the GC–FID system. Koch et al. (54) proposed the separation of MOAH into mono-/ di-aromatic fraction (MDAF) and three/poly aromatic fraction (TPAF) by previously separating the MOSH and MOAH offline, using a silver nitrate loaded silica gel column. The MOAH fraction was then further separated in a donor-acceptor-based HPLC column. Five fractions were pooled together to reach the sensitivity required and to further characterize the TPAF by GC×GC-MS.
Conclusion
The analysis of MOH in food is a challenging task, which requires careful and thoughtful optimization at every step of the analytical procedure, from sample preparation to the analytical determination, and data interpretation. Hyphenated techniques, such as LC–GC, LC–LC–GC, GC×GC, and LC– GC×GC, play a fundamental role in the advances and automation of the analysis. It is the authors’ opinion that MOH determination is among the few applications that benefit from more than two chromatographic dimensions.
Further studies are needed to provide reliable and more detailed data for a full risk-assessment, and it is predicted that hyphenated techniques will once again be
the key players in achieving the level of information required.
Acknowledgements
This work is supported by Fonds de la Recherche Scientifique Belgique (FNRS) (CDR projects-MOHPlatform, J.0170.20). This article is based upon work from the Sample Preparation Task Force and Network, supported by the Division of Analytical Chemistry of the European Chemical Society.
References
1) International Agency for Research on Cancer (IARC), IARC Monogr. Eval. Carcinog. Risks to Humans 100 F (2012).
2) European Food Safety Authorithy (EFSA), EFSA J. 10(6) (2012).
3) M.J. Miller, E.C. Lonardo, R.D. Greer, C. Bevan, D.A. Edwards, J.H. Smith, and J.J Freeman, Regul. Toxicol. Pharmacol. 23(1), 55–68 (1996).
4) L.C. Griffis, L.E. Twerdok, S. Francke‐Carroll, R.W. Biles, R.E. Schroeder, H. Bolte,
H. Faust, W.C. Hall, and J. Rojko, Food Chem. Toxicol. 48(1), 363–372 (2010).
5) P.J. Boogaard, K.O. Goyak, R.W. Biles, L.L.P. van Stee, M.S. Miller, and M.J. Miller, Regul. Toxicol. Pharmacol. 63(1), 69–77 (2012).
6) J. Cravedi, K. Grob, U.C. Nygaard, and J. Alexander, EFSA Support. Publ. 14(2) (2017).
7) L. Barp, M. Biedermann, K. Grob, F. Blas‐Y‐Estrada, U.C. Nygaard, J. Alexander, and J.‐P. Cravedi, Sci. Total Environ. 583, 319–333 (2017).
8) L. Barp, M. Biedermann, K. Grob, F. Blas‐Y‐Estrada, U.C. Nygaard, J. Alexander, and J.‐P. Cravedi, Sci. Total Environ. 575, 1263–1278 (2017).Sci. Total Environ. 575, 1263–1278 (2017).
9) G.W. Trimmer, J.J. Freeman, R.A.J. Priston, and J. Urbanus, Toxicol. Pathol. 32(4), 439–447 (2004).Toxicol. Pathol. 32(4), 439–447 (2004).
10) L. Barp, C. Kornauth, T. Wuerger, M. Rudas, M. Biedermann, A. Reiner, N. Concin, and K.Grob, Food Chem. Toxicol. 72, 312–321 (2014).
11) M. Biedermann, L. Barp, C. Kornauth, T. Würger, M. Rudas, A. Reiner, N. Concin, and K. Grob, Sci. Total Environ. 506–507:644–655 (2015).
12) D. Adenuga, K. Goyak, and R.J. Lewis, Crit. Rev. Toxicol. 47(9), 754–770 (2017).
13) J.‐C. Carrillo, A. van der Wiel, D. Danneels, O. Kral, and P.J. Boogaard, Regul. Toxicol. Pharmacol. 106, 316–333 (2019).
14) European Commission, Off. J. Eur. Union L12(84), 95–96 (2017).
15) S. Bratinova and E. Hoekstra, Joint Research Centre (JRC), Guidance on sampling, analysis and data reporting for the monitoring of mineral oil hydrocarbons in food and food contact materials. In the frame of Commission Recommendation (EU) 2017/84 (2019). doi:10.2760/208879
16) Foodwatch International, project report International test of various canned
baby milk products for their content of mineral oil hydrocarbons (MOSH/MOAH), (October 2019). https://www. foodwatch.org/fileadmin/‐INT/mineral_oil/documents/2019‐10‐24_Projectreport_ babymilk_FINAL.pdf (accessed Feb 3, 2021)
17) D. Arcella, K. Baert, and M. Binaglia, EFSA Support. Publ. 16(11) (2019).
18) European Commission, Off. J. Eur. Union L354(16) (2008).
19) European Commission, Off. J. Eur. Union L12, 1–89 (2011).
20) European Commission, Off. J. Eur. Union L250, 1–84 (2008).
21) European Commission, Off. J. Eur. Union L153(1), 1–186 (2011).
22) Scientific Committee of the Federal Agency for the Safety of the Food Chain, Advice 19‐2017 Action thresholds for mineral oil hydrocarbons in food (2017). at http:// www.afsca.be/scientificcommittee/ opinions/2017/_documents/Advice19‐2017. pdf (accessed Feb 3, 2021).
23) EN European Standard 16995:2017, Foodstuffs - Vegetable oils and foodstuff on basis of vegetable oils - Determination of mineral oil saturated hydrocarbons (MOSH) and mineral oil aromatic hydrocarbons (MOAH) with on-line HPLC-GC-FID analysis (2017).
24) S. Weber, K. Schrag, G. Mildau, T. Kuballa, S.G. Walch, and D.W. Lachenmeier,
Anal. Chem. Insights 13, 1–16 (2018).
25) G. Purcaro, L. Barp, and S. Moret, Anal. Methods 8(29), 5755–5772 (2016).
26) S. Biedermann‐Bremm, K. Grob, Eur. Food Res. Technol. 232(6), 1035–1041 (2011).
27) S. Moret, M. Scolaro, L. Barp, G. Purcaro, M. Sander, and L.S. Conte, Food Chem. 157, 470–475 (2014).
28) R. Lorenzini, K. Fiselier, M. Biedermann, M. Barbanera, I. Braschi, and K. Grob, Food Addit. Contam. – Part A Chem. Anal. Control. Expo. Risk Assess. 27(12), 1765–1774 (2010).
29) M. Biedermann and K. Grob, Eur. Food Res. Technol. 230(5), 785–796 (2010).
30) M. Biedermann and K. Grob, J. Chromatogr. A. 1255, 56–75 (2012).
31) S. Moret, M. Scolaro, L. Barp, G. Purcaro, and L.S. Conte, Food Chem. 196, 50–57 (2016).
32) S. Moret, M. Sander, HG. Purcaro, M. Scolaro, L. Barp, and L.S. Conte,
Talanta 115, 246–252 (2013).
33) S.Moret, L.Barp, K.Grob, and L.S.Conte, Food Chem. 129(4), 1898–1903 (2011).
34) K. Fiselier, F. Grundböck, K. Schön, O. Kappenstein, K. Pfaff, C. Hutzler,
A. Luch, and K. Grob, J. Chromatogr. A. 1271(1), 192–200 (2013).
35) K. Fiselier, D. Fiorini, and K. Grob, Anal. Chim. Acta 634, 96–101 (2009).
36) C. Wagner, H.P. Neukom, V. Galetti, and K. Grob, Mitt Leb. Hyg 92, 231–249 (2001).
37) B. Maurus, F. Katell, and G. Koni, J. Agric. Food Chem. 57(19), 8711–8721 (2009).
38) M. Nestola and T.C. Schmidt, J. Chromatogr. A. 1505, 69–76 (2017).
39) M. Biedermann, C. Munoz, and K. Grob, J. Chromatogr. A. 1624, 461236 (2020).
40) M. Biedermann, C. Munoz, and K. Grob, J. Chromatogr. A. 1521, 140–149 (2017).
41) S.Moret, K.Grob, and L.S.Conte, J. Chromatogr. A. 750(1–2), 361–368 (1996).
42) K. Fiselier, D. Fiorini, and K. Grob, Anal. Chim. Acta 634(1), 102–109 (2009).
43) M. Lommatzsch, M. Biedermann, T.J. Simat, and K. Grob, J. Chromatogr. A. 1402, 94–101 (2015).
44) M. Zoccali, L. Barp, M. Beccaria, D. Sciarrone, G. Purcaro, and L. Mondello,
J. Sep. Sci. 39(3), 623–631 (2016).
45) S.Moret, L.Barp, G.Purcaro, and L.S. Conte, J. Chromatogr. A. 1243, 1–5 (2012).
46) G. Purcaro, P.Q. Tranchida, L. Barp, and S. Moret, Anal. Chim. Acta 773, 97–104 (2013).
47) G. Purcaro, S. Moret, and L. Conte, J. Chromatogr. A. 1255, 100–111 (2012).
48) European Parliament and the Council of the European Union, Off. J. Eur. Communities (L 221/8), 8–36 (2002). doi:10.1017/CBO9781107415324.004
49) M. Biedermann and K. Grob, J. Sep. Sci. 32(21), 3726–3737 (2009).
50) M.Biedermann and K.Grob, J. Chromatogr. A. 1375, 146–153 (2015).
51) S. Pantó, M. Collard, and G. Purcaro, Curr. Trend Mass Spectrom. 18(3), 1–6 (2020).
52) A.R. García‐Cicourel, B. van de Velde, J. Verduin, and H.G. Janssen, J. Chromatogr. A. 1607, 460391 (2019).
53) A. R. García‐Cicourel, B. van de Velde, G. Roskam, and H. G. Janssen, J. Chromatogr. A. 1614, 460713 (2020).
54) M. Koch, E. Becker, M. Päch, S. Kühn, and E. Kirchhoff, J. Sep. Sci. 43(6), 1089–1099 (2020).
Nicola Sdrigotti graduated with honours from the University of Udine, Italy, with an MSc in food quality control.The first approach with analytical chemistry started in the
field of electrochemistry, at his home University. Motivated by the desire for knowledge, he took part in two Erasmus experiences abroad, first in the field of proteomics at the Danish National Food Institute, and then in the field of advanced chromatography for mineral oil analysis at the Gembloux Agro-Bio Tech institute, Belgium. Gregory Bauwens graduated in bioengineering (chemistry orientation) from the University of Liege, Belgium. His master thesis focused on the optimization of a completely hyphenated LC–GC×GC–TOF-MS/FID platform for the analysis of mineral oil. Being very interested in this topic, he has recently started a PhD on the application of advanced analytical techniques for the determination of mineral oil in foods. Giorgia Purcaro has been analytical chemistry professor at the Gembloux Agro Bio Tech Department of the University of Liége (Belgium) since 2018. Her research interests include the development of advanced multidimensional and comprehensive chromatography techniques (GC×GC, LC–GC, LC-GC×GC)
and miniaturized sample preparation approaches for food quality and safety applications. She was awarded the Leslie S. Ettre Award in 2010, and, in 2015, received the J. Philipps award for her contribution to the GC×GC field. She has authored or co-authored over 80 peer-reviewed publications, 10 book chapters, and more than 150 conference presentations.
Direct correspondence to: amatheson@mjhlifesciences.com









New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.
 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")




