A Rapid, Automated Extraction Platform to Assess Drug Product Potency by Online Liquid Chromatography


We present a new automated active pharmaceutical ingredient (API) extraction approach with integrated analysis by online liquid chromatography (LC) to assess drug product potency. The platform consists of an extraction module for repeated dynamic pressurized solvent extractions of tablets. Extractions rely on temperature and pressurized solvents (50:50 0.1% TFA:acetonitrile, 100 bar, 3.0 mL/min) to dissolve the tablet coating (if present), disintegrate the tablet, and disperse and solubilize material. Upon exiting the extraction vessel, solvent carrying extracted material is sampled every 15 s using a dual loop LC injection valve. The valve allows direct injection of drug product extract onto a short LC column for online monitoring of the extraction process and generation of an “extract-o-gram.” Autoprocessing of API peaks in the extract-o-gram facilitated direct calculation of tablet potency by integrating the area under the resulting API concentration-time curve. To assess the performance of the platform, a series of experiments was conducted with four pharmaceutical products spanning a wide API dosage range: 200 mg ibuprofen, 81 mg aspirin, and 12 mg and 1 mg tablets of Lilly development materials (Lilly A and Lilly B). Results were encouraging; using a generic set of extraction conditions, reproducible quantitative recoveries were obtained for ibuprofen (99.3% label claim, 0.5% RSD), aspirin (103.2%, 1.1% RSD), Lilly A (98.5%, 1.1% RSD), and coated and core Lilly B tablets (100.8%, 1.3% RSD and 100.9%, 2.4% RSD, respectively).
Prior to release of pharmaceutical products to the market, materials must meet a set of acceptance criteria that ensure product quality and consistency. Common drug product release tests rely on extraction of the active pharmaceutical ingredient (API) from the dosage unit and subsequent liquid chromatography (LC) analysis, including identity, assay, purity, and uniformity of dosage units. Sample preparation and extraction of an API from a solid oral dosage form is a tedious and labor-intensive activity for many projects in pharmaceutical development. In a typical tablet analysis, the sample preparation step serves to isolate the API from the pharmacologically inactive excipients that comprise the bulk of the sample matrix, while also preparing a solution phase sample for analysis. Traditional sample preparation methods involve transferring the tablet into a volumetric flask, adding large volumes of organic solvent, and either mechanical shaking or sonication of the sample to disintegrate the tablet and extract the API (1,2). Following extraction, sample clarification removes insoluble material in preparation for analysis. Although modern LC methods are relatively fast, sample preparation is often the most time-consuming element of the analysis, comprising up to 80% of the total time (3). Therefore, with the continued trend toward shorter pharmaceutical development timelines using fewer resources, there is always a driver for more efficient sample preparation procedures (4).
To facilitate rapid sample preparation for solid oral dosage forms, several non-traditional sample preparation techniques have been developed. Two common techniques involve the use of elevated temperature, elevated pressure, or particle-size-reduction approaches to increase the efficiency of API solubilization (5). The techniques developed to leverage elevated temperature conditions include accelerated solvent extraction (ASE, also known as a pressurized liquid extraction [PLE]), and microwave assisted extraction (MAE). Elevated temperature and pressure conditions have also been shown to dramatically improve the recovery of extractions while also reducing extraction time (6). Since its development, ASE techniques have been applied effectively to extract several APIs from crushed and whole tablets for both immediate and sustained release solid oral dosage forms (5, 7–12).
Supercritical fluid extractions (SFE) are another ASE-based approach where a super- critical CO2-based fluid, modified with a small amount (<20%) of organic solvent, extracts API from tablet formulations using equipment similar to ASE. SFE has been used extensively in the research setting with tablet dosage forms to improve the environmental impact of the sample preparation process (13–19). The primary limitation to SFE-based extractions is the poor recoveries obtained with polar pharmaceuticals. For example, in the extraction of pseudoephedrine-HCl from tablets up to 20% (v/v) methanol and an ion-pairing reagent (1% v/v, 1-hepatensulfonic acid), modifiers were required to achieve quantitative recoveries (20–22). Subcritical water extractions (SWE) are a subset of PLEs where only aqueous extraction solvents are used. Richter and associates advanced this approach to extract nifedipine from tablet formulations (23), and the Thurbide group more thoroughly characterized the approach using acetaminophen, vitamin C, and fluoxetine-HCl from tablet and capsule formulations at extraction temperatures up to 225 °C (24,25). Most notably, quantitative recoveries were observed without thermal decomposition of the API at such temperatures. Microwave-assisted extractions (MAE) have also garnered interest as an alternative extraction technique, because of the relative simplicity of the instrument and the highly parallel extraction potential of the systems.
The two most used particle-size-reduction approaches to extraction are ball milling and homogenization. Both techniques act to reduce sample particle size, increasing sample surface area and leading to increased extraction efficiency (5). Ball milling pulverizes samples in the presence of the extraction solvent using stainless steel balls. Following extraction, centrifugation is used to separate insoluble material prior to analysis. Kok and Debets (26) showed that milling and extraction times on the order of minutes could be performed for a variety of high- and low-dose tablets. While relatively fast, ball milling requires several manual manipulations, and is limited in the amount of extraction solvent that can be used. Homogenization uses a set of rotating blades combined with wet grinding, shredding, or shearing to reduce sample particle size and increase surface area for rapid sample disintegration (27). The primary advantage to the homogenization approach for tablet formulations is its current integration into an automated robotic sample preparation system known as the Tablet Processing Workstation II (Sotax) (5,28–30). Shamrock and associates (31) used the automated sample preparation work- flow to transfer a manual sample workflow for roxifiban tablets to an automated whole tablet workflow, where samples were automatically weighed, disintegrated by homogenization, extracted, automatically diluted, and transferred to LC autosampler vials. When paired with effective automation systems, homogenization facilitates rapid sample preparation for solid oral dosage formulations, but the cost and complexity of this instrumentation is a limitation.
In this work, we describe a new automated API extraction platform with integrated analysis by online LC to assess drug product potency for solid oral dosage forms. The platform includes an extraction module for repeated dynamic pressurized liquid extractions of drug product tablets. Extractions rely on a generic set of extraction conditions to dissolve the tablet coating (if present), disintegrate the tablet, and then disperse and solubilize the API. Upon exiting the vessel, extraction solvent travels to a dual loop LC injection valve for online, quantitative monitoring of the extraction process. To assess the performance of the assay, we conducted a series of proof-of-concept experiments with four drug product samples—two over-the-counter products and two Eli Lilly and Company development materials.
Experimental
Instrumentation
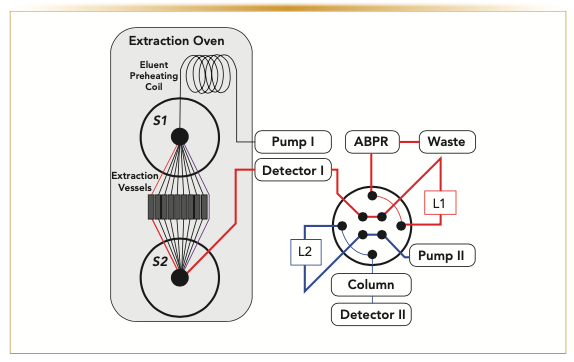
Figure 1 shows a diagram of the instrument used to automate tablet potency analysis. A 30 x 10 mm i.d., 2-μm frit porosity, modular high-performance LC (HPLC) column hardware (IDEX H&S) served as the extraction vessel, allowing tablet disintegration and extraction while preventing insoluble material from traveling to downstream fluidic components. Total volume for the extraction vessels was 2.35 mL. Extraction vessels were inside a Waters MV-10 analytical supercritical fluid extraction system oven (Waters Corporation). An Agilent 1260 analytical scale binary pump (G1312B, Agilent Technologies) delivered extraction solvent to each vessel. Extraction fluid temperature was equilibrated at the extraction temperature while passing through a 5 mL sample loop (Rheodyne). Residence time in the preheating coil was 100 s. Detector I was an in-line UV absorbance detector (Agilent 1260, G1314B); the detector was connected directly to the outlet of the extraction oven. To prevent bursting the inline flow cell at extraction pressures above 100 bar, the detector was fitted with a high-pressure flow cell (G1314-60082; 10 mm pathlength; V = 14 μL; Pmax = 400 bar). The outlet of the flow cell was connected to an Agilent 8-port two-position duo valve (P/N 5067-4244) using a 5-foot coil of 0.040” i.d. stainless steel tubing (IDEX). The valve had two 1.2- μL injection loops (L1 and L2) for direct injection of the material exiting the extraction vessels onto the chromatographic column. The column was in an Agilent 1290 thermostated column compartment (G1316). The waste port of the valve was connected to the MV-10 active back pressure regulator (ABPR) using a second 5-foot coil of 0.040” i.d. stainless steel tubing (IDEX). Pump II, an Agilent 1290 qua- ternary pump (G4204A), delivered mobile phase to the separation column. The outlet of the column was connected to Detector II, an Agilent 1290 DAD (G4212B) equipped with a 0.4 μL flow cell (G4212-60032; 3.7 mm pathlength). The instrument was controlled using software from both vendors Agilent ChemStation (OpenLab C.01.07) and Waters ChromScope (Version 1.2).

 placed inside the extraction oven directed flow to up to nine individual extraction vessels. Port 10 in both valves provided an extraction vessel bypass (purple). An eight-port two-position valve was equipped with dual 1.2 μL sample loops (L1 and L2) to interface the extraction with the online LC. Detectors I and II were both ultraviolet (UV) absorbance detectors.")
FIGURE 1: Instrument configuration schematic for automated drug product extractions. The instrument consisted of two independent LC pumps; Pump I performed the dynamic PLE; Pump II was connected to the LC column. Two 10-port column selection valves (S1 and S2) placed inside the extraction oven directed flow to up to nine individual extraction vessels. Port 10 in both valves provided an extraction vessel bypass (purple). An eight-port two-position valve was equipped with dual 1.2 μL sample loops (L1 and L2) to interface the extraction with the online LC. Detectors I and II were both ultraviolet (UV) absorbance detectors.
Chemicals and Reagents
Aspirin and ibuprofen were from Sigma Aldrich; Lilly’s Corporate Reference Standards Organization provided standard samples for the two Lilly development materials studied—development material A (Lilly A) and development material B (Lilly B). Acetonitrile (LC–MS Optima grade) and premade 0.1% trifluoroacetic acid (TFA) in water were from Fisher Scientific and EMD Millipore, respectively. Method development and calibration standards for each solute were made in 50:50 acetonitrile:0.1% TFA in water.
Extraction Conditions
Drug product extractions utilized a dynamic extraction mode with a genericized set of conditions for all proof-of-concept work. Extraction fluid was pumped through the extraction vessels for 25 min at 3.0 mL/min. Extraction solvent consisted of 50:50 acetonitrile:0.1% TFA in water; extraction oven temperature was 30 °C. The ABPR pressure setting was 100 bar.
Chromatographic Conditions
All separations used an Agilent Zorbax SB-C18 (50 mm x 3.0 mm i.d., 1.8 μm dp) column and an acetonitrile:0.1% TFA in water mobile phase. Acetonitrile composition for each solute was set to achieve a retention time of about 12 s. Acetonitrile compositions for ibuprofen, aspirin, Lilly A, and Lilly B were 0.65, 0.25, 0.275, and 0.30, respectively. Injection-to-injection cycle time, (valve switching time or chromatographic time) was 15 s. The extract-o-gram run time was 25 min. In total, 100 LC injections were performed for each extraction experiment. Injection volume, column temperature and mobile phase flow rate for all experiments were 1.2 μL, 85 °C, and 2.5 mL/min, respectively; Interstitial linear velocity was about 1.0 cm/s. Detection at 80 Hz used absorbance at 220, 254, and 280 nm. Quantitation used the wavelength with the best signal for the specific sample. Peaks were autointegrated in the chromatographic software, and quantified by response factor from a single point calibration. Standard concentration was selected to fall within the linear range of the detector, and be relevant to the tablet dosage for each product evaluated.
Results and Discussion
Automation of Drug Product Potency Workflow by Fast, Online LC
Figure 1 shows a block diagram for the instrument designed to automate the tablet potency assay for up to nine replicate drug product samples. Briefly, the platform consisted of an extraction module for repeated PLE of tablets in 2.35 mL extraction vessels. Extractions utilized temperature and pressurized acetonitrile:water mixtures to dissolve the tablet coating (if present), disintegrate the tablet, and disperse and solubilize material during the 25 min extraction time. Extraction solvent composition was selected to have good solubility for the APIs tested. Online LC monitored the dynamic extraction process using the dual loop injection valve with high temporal resolution sampling, 15 s sampling cycle, and chromatographic analysis. High frequency sampling and analysis were key to quantitative performance.
Figure 2 shows representative data from an extraction experiment, highlighting platform performance and utilization of online LC for quantitation. Drug product content determination began by manually loading the extraction vessels with the desired sample and placing extraction vessels in the extraction oven. Each PLE was initiated when, as shown in Figure 1, S1 and S2 valves rotated to place the extraction vessel into the flow path of the extraction solvent. Online LC analysis start time was aligned with this extraction start time beginning the repeated actuation of the dual loop injection valve every 15 s. Figure 2a shows the pressure profile obtained at the extraction pump (Pump 1). Each PLE can be broken down into three distinct phases based on this profile, a filling phase, shown in red, where the empty extraction vessel filled with solvent and was pressurized by the ABPR to the desired extraction pressure (100 bar); note that the instrument contributed approximately 50 bar to the extraction pump, accounting for the offset in pressure readings between the extraction pump and ABPR setpoint. Following pressurization, the extraction entered a disintegration phase, shown in blue, where the tablet coating was penetrated by the solvent breaking up the tablet, and the solvent solubilized the API. Completion of the disintegration phase was determined by detection of the first API peak in the online LC chromatogram of Figure 2b, 3.75 minutes into the extract-o-gram. The first several minutes of the extract-o-gram correspond to the filling and disintegration phases. During these phases, the two-position valve switched every 15 s injecting extraction solvent onto the online LC column. Each 15 s cycle represents a single chromatographic analysis; conditions were optimized so API peaks were eluted with a retention time of ~12 s to enable the subsequent injection. Following the disintegration phase, the extraction entered the steady-state phase, shown in green in Figure 2a. During the steady-state phase, API concentration in the extraction solvent increased, and a commensurate increase in API peak size (red arrows in Figure 2b and profile in 2c) in the chromatograms of the extract-o-gram was observed. Figure 2c shows the maximum concentration of API in the extraction solvent was observed at 8 min, and then fell off as the extraction completed its quantitative removal of API from the drug product matrix.

FIGURE 2: Example experimental data stream used in the automated tablet potency workflow. (a) Pump I pressure profile collected during the extraction. During the filling phase, pump I filled the vessel with solvent, and in combination with the ABPR, pressurized the vessel to the desired extraction pressure. Following pressurization, the vessel entered the disintegration phase where extraction pressure stabilized, and solvent penetrated the tablet coating (if present). The steady-state phase represents the time where the vessel maintains set pressure and temperature for the duration the extraction; (b) Chromatograms observed at detector II during the initial five minutes of the drug product extraction. Red and blue dashed lines indicate sampling valve actuations, that is, every 15 s; color highlights the dual injection loop-sampling scheme used; (c) Representative 25-min extract-o-gram generated when using online LC to monitor the extraction of a drug product with 15 s time resolution. Elution conditions were optimized for each API peak retention time to be ~12 s.

Auto-integration and comparison to the external standard of the 100 individual online LC injections collected during the extraction resulted in an API concentration value, Ci, for each sample in the extract-o-gram. Equation 1 was used to determine the mass of API, mi, recovered from each drug product extraction:


where Ci is the sample concentration in each online LC chromatogram, Fe is the extraction flow rate, t is extraction time (25 min), and dt is the online LC sampling time (15 s). Multiplication of the sample concentration Ci (mg/mL) by Fe (3.0 mL/min) resulted in a value for API in mass per time for each LC injection. Multiplication of the mass per time in each online LC chromatogram by the 15 s sampling time resulted in the mass of API, mi, in each extract-o-gram chromatogram. Integration of the mass values during the entirety of the extraction yielded a total mass of API recovered during the extraction.
Over-the-Counter Extraction Examples
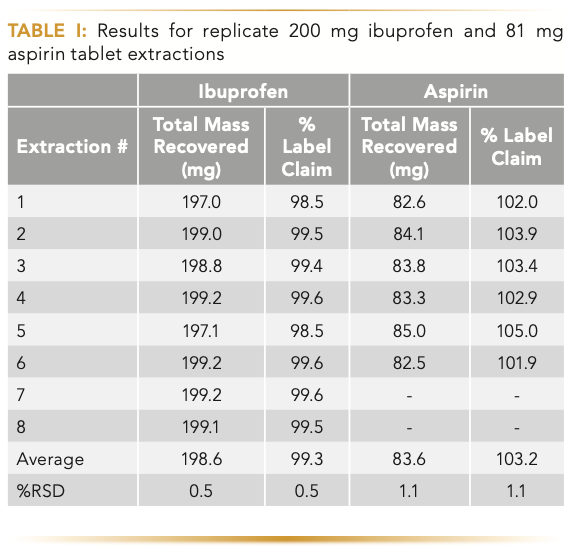
Performance of the extraction platform was assessed using proof-of-concept extractions with two over-the-counter (OTC) materials. Figure 2c shows a representative extraction profile for one of the two OTC materials tested: 200 mg ibuprofen immediate release tablets and 81 mg delayed release aspirin tablets. Results for both OTC materials were encouraging. Table I shows the total mass recovered, and percent label claim for eight ibuprofen and six aspirin extractions. For ibuprofen, an average of 198.6 mg or 99.3% of label claim was recovered, with a 0.5% RSD and 83.6 mg or 103.2% of label claim for aspirin was recovered with a 1.1% RSD. Recovery and reproducibility were quite good for both materials, conforming to the USP requirements for drug product potency (90–110% of labeled claim on content, USP <905> Uniformity of Dosage Units).

Application to Lilly Development Materials
Figure 3 shows representative extract-o-grams for two Lilly development materials, Lilly A, and Lilly B (coated and uncoated tablets), and the quantitative results from the three series of six replicate extractions reported as percent label claim. Figure 3a shows an example extract-o-gram for 12 mg coated immediate release tablets of Lilly A. The inset chromatogram was pulled from 9.75–10.00 min into the extraction, a time corresponding to that required to extract greater than 95% of Lilly A from the tablet. An average of 98.5% of label claim material was recovered from Lilly A tablets with an RSD of 1.2%. Figures 3b and 3c show example extract-o-grams for low dose 1 mg uncoated tablet cores and 1 mg coated tablets of Lilly B. Extraction results for the two low dose materials were the same, with the average API mass recovered from both materials being 1.01 mg with RSD of 1.3% and 2.4% for the core and coated tablets, respectively. A qualitative comparison of the core and coated extract-o-grams showed similar extraction profiles for the two samples. Close inspection highlights that the concentration of Lilly B for the tablet core extraction is marginally higher than the coated tablets early in the extraction, as the tablet coating present must be penetrated by the solvent prior to observation of Lilly B in the extraction solvent.

FIGURE 3: Representative extraction profiles for three development materials evaluated. (a) Extraction profile for a 12 mg tablet of Lilly A. The inset chromatogram was from 9.75-10.00 minutes into the extraction, corresponding to the time required to extract greater than 95% of the API from the tablet matrix; (b) Example profile from the extraction of a 1 mg uncoated tablet core of Lilly B; (c) Extraction profile from a coated 1 mg tablet of Lilly B; (d) Quantitative extraction results from Lilly A and Lilly B as a percent recovery based off of label claim.

Conclusions
A new automated API extraction platform with integrated analysis by online liquid chromatography was developed to determine drug product potency. Using a generic set of extraction conditions, proof-of-concept experiments were performed on four different drug products: 200 mg ibuprofen, 83 mg aspirin tablets, and two Lilly development materials. Quantitative recoveries of 98.5% to 103.2% were obtained for tablets containing 1 to 200 mg API. Reproducibility was also quite good, with extraction RSD for all materials evaluated being less than 2.5%.
Notes
The authors declare no competing financial interest.
Acknowledgments
Funding for this work was provided by a Lilly Research Labs Process Research and Development Post-Doctoral Fellowship. We would like to thank Waters Corporation for the loan of instrumentation.
References
(1) B. Nickerson, Sample Preparation of Pharmaceutical Dosage Forms (Springer, New York, New York, 2011). https://doi.org/10.1007/978-1-4419-9631-2.
(2) C.K. Cho and M.W. Dong, Sep. Sci. Tech. 6, 123–144 (2005). https://doi.org/10.1016/S0149-6395(05)80049-2.
(3) H. Kataoka, TrAC Trend Anal. Chem. 22(4), 232–244 (2003). https://doi.org/10.1016/S0165-9936(03)00402-3.
(4) A.M. Pimenta, M.C.B.S.M. Montenegro, A.N. Araújo, and J.M. Calatayud, J. Pharmaceut. Biomed. 40(1), 16–34 (2006). https://doi.org/10.1016/j.jpba.2005.10.006.
(5) B. Nickerson, W.B. Arikpo, M.R. Berry, V.J. Bobin, T.L. Houck, H.L. Mansour, and J. Warzeka, J. Pharmaceut. Biomed. 47(2), 268–278 (2008). https://doi.org/10.1016/j.jpba.2008.01.006.
(6) B.E. Richter, B.A. Jones, J.L. Ezzell, N.L. Porter, N. Avdalovic, and C. Pohl, Anal. Chem. 68(6), 1033–1039 (1996). https://doi.org/10.1021/ac9508199.
(7) A. Blanchard, C. Lee, B. Nickerson, L.L. Lohr, A.J. Jensen, K.M. Alsante, T.R. Sharp, D.P. Santafianos, R. Morris, and K.D. Snyder, J. Pharmaceut. Biomed. 36(2), 265–275 (2004). https://doi.org/10.1016/j.jpba.2004.05.012.
(8) A.M. Abend, L. Chung, D.G. McCol- lum, and W.P. Wuelfing, J. Pharmaceut. Biomed. 31(6), 1177–1183 (2003). https://doi.org/10.1016/S0731-7085(03)00020-7.
(9) E. Björklund, M. Järemo, L. Mathiasson, L. Karlsson, J.T. Strode, J. Eriksson, and A. Torstensson, J. Liq. Chromatogr. Relat. Technol. 21(4), 535–549 (1998). https://doi.org/10.1080/10826079808001238.
(10) T.H. Hoang, R. Farkas, C. Wells, S. McClintock, and M. Di Maso, J. Chromatogr. A 968(1), 257–261 (2002). https://doi.org/10.1016/S0021-9673(02)00956-1.
(11) C. Lee, J. Gallo, W. Arikpo, and V. Bobin, J. Pharmaceut. Biomed. 45(4), 565–571 (2007). https://doi.org/10.1002/jps.2600831104.
(12) F.L. Alkhateeb, K.B. Thurbide, Anal. Methods 7(4), 1509–1516 (2015). https://doi.org/10.1039/C4AY02876G.
(13) A.L. Howard, M.C. Shah, D.P. Ip, M.A. Brooks, J. Thompson Strode, and L.T. Taylor, J. Pharm. Sci. 83(11), 1537–1542 (1994). https://doi.org/10.1002/jps.2600831104.
(14) B.R. Simmons, O. Chukwumerije, and J.T. Stewart, J. Pharmaceut. Biomed. 16(3), 395-403 (1997). https://doi.org/10.1016/S0731-7085(97)00093-9.
(15) J.K. Lawrence, A.K. Larsen, and I.R. Tebbett, Anal. Chim. Acta 288(1), 123–130 (1994). https://doi.org/10.1016/0003-2670(94)85121-2.
(16) N. Bahramifar, Y. Yamini, and M. Shamsipur, J. Supercrit. Fluids 35(3), 205–211 (2005). https://doi.org/10.1016/j.supflu.2005.01.005.
(17) J.R. Dean and J. Lowdon, Analyst 118(7), 747–751 (1993). https://doi.org/10.1039/AN9931800747.
(18) S. Scalia, G. Ruberto, and F. Bonina, J. Pharm. Sci. 84(4), 433–436 (1995). https://doi.org/10.1002/jps.2600840409.
(19) P.R. Eckard, J.T.B. Strode, L.T. Taylor, A.L. Howard, and D. Ip, J. Chromatogr. Sci. 36(3), 139–145 (1998). https://doi.org/10.1093/chromsci/36.3.139.
(20) M. Ashraf-Khorassani, S. Hinman, and L.T. Taylor, J. High Res. Chromatog. 22(5), 271–275 (1999). https://doi.org/10.1002/(SICI)1521-4168(19990501)22:5.
(21) P.R. Eckard and L.T. Taylor, J. Pharmaceut. Biomed. 15(5), 613–619. https://doi.org/10.1016/S0731-7085(96)01897-3.
(22) P.R. Eckard and L.T. Taylor, J. High Res. Chromatogr. 22(8), 469–474 (1999). https://doi.org/10.1002/(SICI)1521-4168(19990801)22:8.
(23) P. Richter, M.I. Toral, C. Toledo, J. AOAC Int. 89(2), 365–368 (2006). https://doi.org/10.1093/jaoac/89.2.365.
(24) J.N. Murakami and K.B. Thurbide, Can. J. Chem. 92(1), 26–32 (2013). https://doi.org/10.1139/cjc-2013-0411.
(25) J.N. Murakami, K.B. Thurbide, G. Lambertus, and E. Jensen, J. Chromatogr. A 1250, 80–84 (2012). https://doi.org/10.1016/j.chroma.2012.04.026.
(26) S.J. Kok and A.J.J. Debets, J. Pharmaceut. Biomed. 26(4), 599–604 (2001). https://doi.org/10.1016/S0731-7085(01)00488-5.
(27) S. Ahuja and M. Dong, Handbook of Pharmaceutical Analysis by HPLC, Vol. 6 (Elsevier, San Diego, California, 2005).
(28) I. Toro, J.F. Dulsat, J.L. Fábregas, and J. Claramunt, J. Pharmaceut. Biomed. 36(1), 57–63 (2004). https://doi.org/10.1016/j.jpba.2004.05.011.
(29) U. Höller, C. Brodhag, A. Knöbel, P. Hofmann, and V. Spitzer, J. Pharmaceut. Biomed. 31(1), 151–158 (2003). https://doi.org/10.1016/S0731-7085(02)00574-5.
(30) S.M. Han and A. Munro, J. Pharmaceut. Biomed. 20(5), 785–790 (1999). https://doi.org/10.1016/S0731-7085(99)00078-3.
(31) W.F. Shamrock, K. Reilly, and D.K. Lloyd, J. Pharmaceut. Biomed. 21(6), 1225–1232 (2000). https://doi.org/10.1016/S0731-7085(99)00232-0.
About The Authors

Stephen Groskreutz is a senior research scientist in Synthetic Molecule Design and Development at Eli Lilly and Company, Indianapolis, Indiana.


Gordon Lambertus is a research advisor and group leader in the Synthetic Molecule Design and Development at Eli Lilly and Company, in Indianapolis, Indiana.

About The Column Editor

David S. Bell is a Research Fellow at Restek Corporation. He also serves on the Editorial Advisory Board for LCGC and is the Editor for “Column Watch.” Over the past 20+ years, he has worked directly in the chromatography industry, focusing his efforts on the design, development, and application of chromatographic stationary phases to advance gas chromatography, liquid chromatography, and related hyphenated techniques. His main objectives have been to create and promote novel separation technologies and to conduct research on molecular interactions that contribute to retention and selectivity in an array of chromatographic processes. His research results have been presented in symposia worldwide, and have resulted in numerous peer-reviewed journal and trade magazine articles. Direct correspondence to: LCGCedit@mmhgroup.com

















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).



