What Are My Options to Improve My Separation? Part 2: Likelihood of Separation, Adjusting Selectivity for Simple Mixtures
When high performance liquid chromatography (HPLC) users are confronted with questions about how to improve upon the performance of an existing or recently developed method, there are many technological options to consider. This series offers guidance for a systematic approach to method improvement that can save time and resources and likely produce more robust methods. Part 2 explains options adjusting selectivity (peak spacing or elution pattern) when dealing with relatively simple mixtures—which are not always easy to separate—with a focus on reversed-phase LC separations.
As explained in a recent article (1), this article series was prompted by a question I was asked during a visit to a local laboratory here in Minnesota, USA. The question was essentially, “What are my options for improving an old high performance liquid chromatography (HPLC) method that works, but probably is not ideal?” There are many options, but it can be difficult to know where to start. This series of articles is intended to make it more manageable to think about how to proceed. Not all the options we will discuss will be available to all users, but it is valuable to understand what the full array of options looks like, so that you can feel confident and make an informed decision about how to proceed.


High performance liquid chromatography HPLC | Image Credit: © Sophie - stock.adobe.com

In last month’s instalment of “LC Troubleshooting”, I kicked off this series of articles by reviewing and discussing some foundational concepts that form the basis of a systematic approach to thinking about how to improve the resolution (and, indirectly, speed) of a separation. I discussed how different separation challenges (for example, simple vs. complex mixtures) can require different approaches to improving resolution, and talked through a menu of options to improve performance, including both kinetic (that is, factors affecting peak width) and thermodynamic (that is, factors that affect peak speak spacing) factors. I also discussed the importance of resolution as a metric of separation performance because it is a measure of peak separation that quantitatively takes into account both the widths of peaks and the distance between them (that is, the distance between the apices of the peaks). Finally, I discussed the influence of different factors on resolution, including retention (as measured by retention factor, k), efficiency (that is, plate number, N), and selectivity (α). The relationship between resolution and these factors is often examined using the Purnell equation, and one of the key takeaways is that the influence of the selectivity of resolution is greater over a large range than the influences of retention and efficiency. Thus, it is always wise to think about how one might change selectivity as a means of improving resolution.
In the second instalment of this series, I focus on options to improve the performance of separations of relatively simple mixtures. In this case, because it is usually possible to fully resolve the components of the mixture, we can also think about how to improve the speed (throughput) of the separation; however, our discussion of speed will be addressed in a future instalment. Many of the details of this discussion are applicable to the specific case of reversed-phase LC separations.
Defining a “Simple Mixture”
For the purposes of discussion in this series of articles, I think of a “simple mixture” as one containing a small number of compounds. What constitutes a number that is small enough for the mixture to be considered simple is discussed in the next section. In a very rough sense, I always consider a mixture with fewer than five components to be simple, and a mixture with more than 100 components to be complex. What happens between five and 100 really depends on the chemistry of the analytes and the goals of the separation. Readers interested in this aspect are referred to the discussion of “regular” and “irregular” samples by Snyder and Dolan (2). Although this is also a useful framework for thinking about how to approach method development, we will not discuss this framework here. It is very important to note here that just because I refer to a mixture as “simple” does not mean it will be easy to separate. Indeed, a mixture of just two analytes can be very difficult to separate, as in the case of a pair of enantiomers. In most cases, it will be impossible to resolve such a pair of compounds, unless a chiral stationary phase is involved.
What Are My Chances of Separating the Mixture I Have?
One way to answer this question about the probability of separating a particular mixture is to use the statistical overlap theory (SOT), which was developed mainly by Davis and Giddings in 1983, with their seminal paper introducing the idea (3). The SOT is based on a number of assumptions that may not be strictly consistent with the characteristics of a real method development problem; however, I think it is still a very useful means of thinking through the chances of actually separating a particular mixture at hand. If SOT predicts that the probability of separating the mixture is less than 1%, then it would probably be a major waste of time and resources to try to fully resolve the mixture, and we would be better off thinking about alternatives, such as two-dimensional (2D) separations or multiple methods (see below for more on this). One of the major assumptions of SOT is that peaks are randomly distributed throughout the resulting chromatogram when a mixture is separated. For a diverse mixture of analytes, the distribution of peaks is usually close enough to random for SOT to be applicable, but there certainly are exceptions to this. For example, separations of homologous series of analytes (for example, alkanes and alkylbenzenes) produce chromatograms with highly ordered patterns of peaks. SOT cannot reasonably be applied to separations of samples that are dominated by compounds like this. In our discussion here, we only scratch the surface of SOT; readers interested in learning more are referred to the original paper by Davis and Giddings (3), and the subsequent literature that refers to this early work (4).
When considering simple mixtures, we are usually most interested in fully resolving all the components of the mixture. In SOT, this outcome is given the symbol P1m, where m is the number of components in the sample, and nc is the peak capacity of the separation:

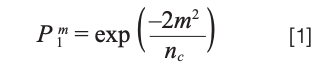
Conceptually, peak capacity quantifies the number of peaks that can fit perfectly side-by-side in a given separation space without overlapping. Under isocratic conditions, this can be calculated easily from the desired resolution between adjacent peaks (RS), the plate number (N), and the retention factor of the most retained component in the separation (kn), as shown in equation 2:

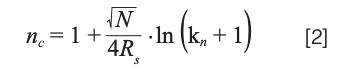
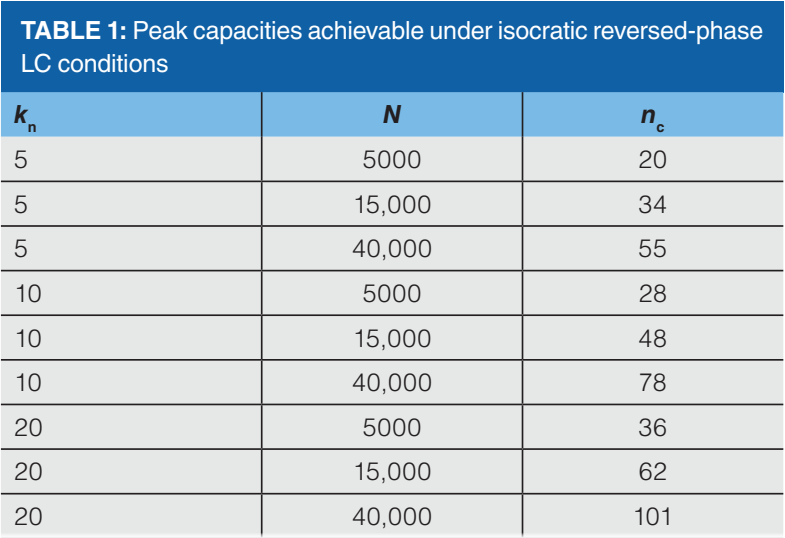
Most calculations of peak capacity in the literature involve a resolution of 1, but if we want to baseline resolve them for quantitative purposes, then we need at least 1.5, and I have used that number here. Table 1 shows the peak capacities calculated using equation 2 for some combinations of kn and N that are relevant in everyday practice of isocratic reversed-phase LC. The kn values span a range from short (5) to long (20) separations, and the plate numbers span a range from what can be achieved easily on a short column with almost any instrument (5000), to what can only be achieved with modern columns and high-performing instruments (40000). We see that the range of peak capacities that results is approximately 20 to 100. Realizing a peak capacity beyond 100 requires gradient elution conditions, 2D separations, or both; these are topics we will address in future instalments in this series.

Now that we have a sense for the range in peak capacities that can be expected under a typical array of conditions, we can turn back to the original question: What is the probability of separating a given mixture with these peak capacities? Figure 1 shows this probability for mixtures in the range of 2–50 components, and peak capacities of 20, 100, 300, or 1000.

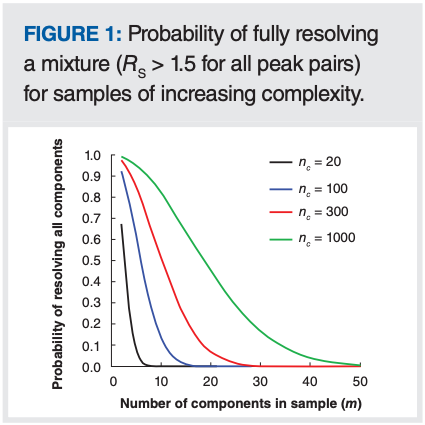
There are a number of important takeaways we can glean from this result. First, with a peak capacity of 20, the probability of resolving even a relatively simple five-component mixture is just 8%. This low probability is a harsh reality of statistics applied to chromatography. It is absolutely true that we can significantly improve these odds by adjusting the mobile phase chemistry, stationary phase chemistry, or both, to reorganize the peak pattern, but in the absence of this effort, the likelihood of fully resolving the mixture on the first try using a short separation and modest column are disappointingly low. In the process of method development, we should never be surprised if we cannot resolve our mixture on the first go. Second, when using a higher peak capacity separation of 100, we see that the probability of resolving the five-component mixture increases dramatically to 60%. This is still far from a “sure thing”, but at least now the odds of success on the first try are better than 50%. Now, if we follow the blue line to the right, we see that the probability drops rapidly, reaching 1% for a 16-component mixture. Beyond 16 components, the likelihood of fully resolving the mixture is vanishingly small, and it will take a great deal of effort in method development to find conditions that provide complete resolution of the mixture. Finally, we see that improving peak capacity to 300 (achievable with a single reversed-phase LC column, under gradient conditions) improves things significantly, moving the 1% mark out to a 27-component sample. Giving ourselves a reasonable chance of resolving mixtures more complex than this requires even higher peak capacities, which can only be provided by 2D separations in most cases, at least under practically useful conditions.
An alternative to 2D separation that is sometimes used for samples of intermediate complexity is to use multiple conventional LC methods to target and resolve different subsets of the components of a mixture. For example, a C18 column might be used to resolve components 1–12, and a phenyl column may be used to separate components 13–20. The information provided by the two methods run separately for each sample can then be combined to provide the information needed for all 20 analytes in the mixture, whereas developing a single method to yield the same information may be very difficult and require a great deal of method development time and resources.
Improving Resolution Through Thermodynamic Adjustments: Changing the Selectivity
As discussed in the preceding section, the likelihood of separating even relatively simple mixtures using a single column under isocratic conditions is dismally low. Fortunately, we have at our disposal a number of levers we can use in method development to overcome the harsh reality of the statistics of random distributions and reorganize peaks to make better use of the space in our chromatograms. In LC, the chemistries of both the mobile and stationary phases can have significant impacts on selectivity, and thus the distribution of peaks in a chromatogram. In general, changing the mobile phase is easier experimentally, but has less of an impact on selectivity; changing the stationary phase is more expensive and harder (but not impossible) to automate, but has more potential for providing large changes in selectivity.
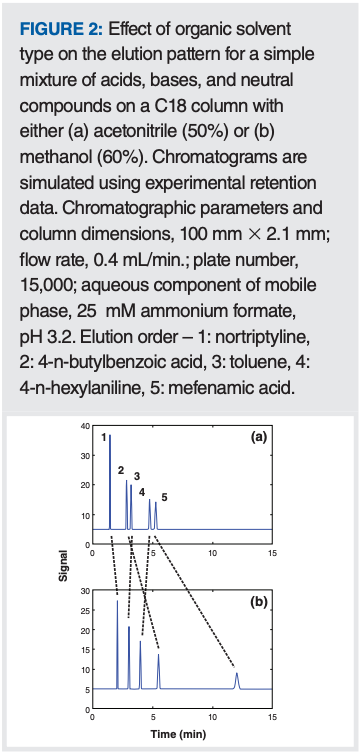
Adjusting the Mobile Phase: When it comes to adjusting the mobile phase, in reversed-phase LC, both the solvent type and the aqueous or organic solvent composition influence selectivity. Currently, we do not know how to predict which organic solvent will provide the most useful selectivity for a given mixture of molecules, thus it is most common in practice to simply do a solvent screen early in method development to see which solvent is most likely to provide the needed selectivity. Figure 2 shows that the impact of the solvent type on the peak pattern can be significant. The dashed lines connecting the peaks for specific compounds are drawn in to indicate which compounds are impacted the most, and that elution order reversals can occur.

We also do not know how to predict a priori the amount of organic solvent (that is, the organic:aqueous ratio) in the mobile phase that will yield the selectivity needed for a successful separation. However, the good news is that once a solvent is chosen, the retention behaviour for a given molecule is consistent enough that a retention model can be built from a few initial retention measurements. With this model in hand, optimal conditions can be found by simulation and then verified by experiment. The development of this general approach began in the 1980s with the work of Snyder and Dolan and coworkers (5), and the approach has enjoyed great success through the years, with development of several commercially available software packages along the way (such as DryLab and LC Simulator). Figure 3(a) shows an example of how a retention model can identify mobile phase compositions that yield selectivities that looks promising (see dashed lines C and D) to fully resolve the mixture. Figure 3(b–d) show chromatograms simulated using mobile phase compositions highlighted by the dashed lines in the plot. In Figure 3(b) we see coelution of mefenamic acid and 4-n-hexylaniline, which is predicted by the crossing of the green and purple lines in Figure 3(a). In Figures 3(c) and (d), we see that the mixture is fully resolved, but the conditions in (d) are more attractive because they yield resolution of the mixture in a shorter time.

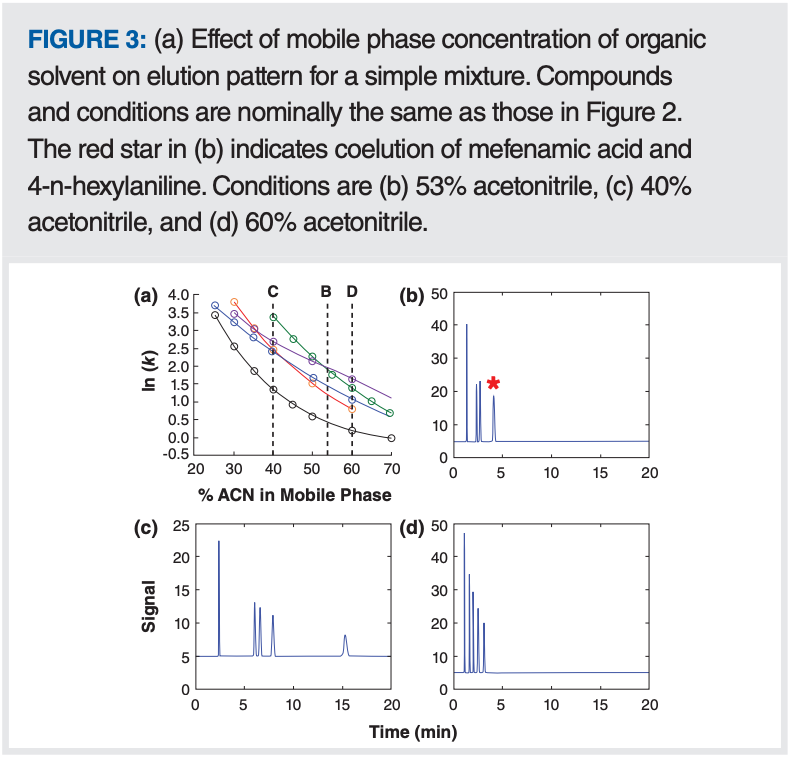
Using this approach to improve isocratic separations is straightforward in practice and requires a few initial “training experiments” where the mobile phase composition is deliberately varied so that the dependence of retention on mobile phase composition can be established. These data can then be fed into the software that allows identification of candidate conditions that are likely to provide resolution of the sample mixture in the shortest analysis time.
In addition to changing the organic solvent type and amount (as shown in Figures 2 and 3), changing the mobile phase pH can also be a powerful way to adjust selectivity, particularly when the sample mixture contains ionogenic analytes. This is a complex topic that requires too much discussion to address here; however, it has been covered extensively in previous instalments of “LC Troubleshooting”; interested readers are referred to those prior instalments to learn more (6).
Adjusting the Stationary Phase: Changing the stationary phase can also be an effective means of adjusting selectivity. As I often tell my students, the good news is that we have approximately 1000 different reversed-phase LC columns to choose from in the marketplace. The bad news is that in practical method development, we don’t have the time or resources to try them all, so we have to choose a few to try. Figure 4 shows an example of the dramatic impact that changing the stationary phase can have on the elution pattern for a simple mixture. The extent to which the changes are more or less different than the change observed when changing the organic solvent type (as in Figure 2) will depend on the chemistry of the analytes and stationary phases involved. Our ability to predict which stationary phases are the right ones to try is pretty limited, but there are some tools to help with this.

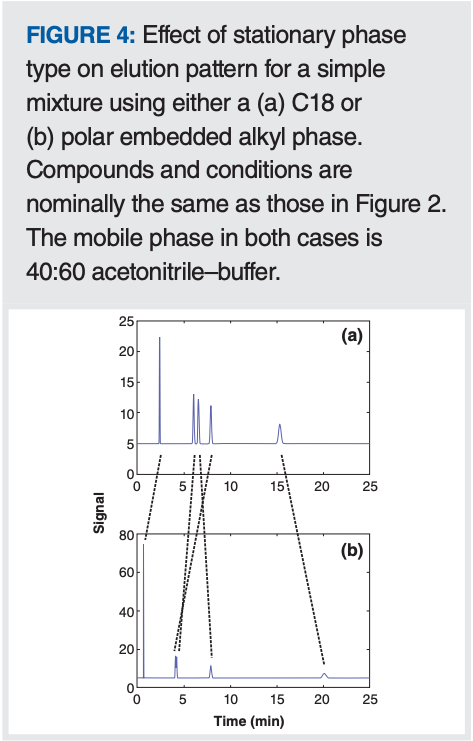
The hydrophobic subtraction model (HSM) of reversed-phase selectivity, developed by Snyder and Dolan and coworkers (7), provides a means of identifying column pairs that are either similar or different. In the context of method development where we want to adjust selectivity by changing the stationary phase chemistry, the HSM can be used to suggest candidate columns to try. Currently, data for approximately 750 columns are available and free web-based tools can be used to interrogate the results. Readers interested in learning more about the model and use of the tools are referred to previous articles in LCGC (8), and the websites hosting the tools (https://apps.usp.org/app/USPNF/columnsDB.html and https://www.hplccolumns.org/).
Summary
Many HPLC users are confronted with questions about how to improve upon the performance of an existing method. Fortunately, modern HPLC technologies (that is, instrumentation, columns, and software) provide many possible avenues to make improvements. However, the number of possibilities can be intimidating, making it difficult to decide upon a particular path to improvement. In this instalment of “LC Troubleshooting”, I have discussed options for adjusting selectivity (that is, peak spacing or elution pattern) when dealing with relatively simple mixtures, with a focus on reversed-phase LC separations. In LC, both the chemistry of the mobile phase and the stationary phase strongly influence selectivity, thus changing one or both of them can be a viable means of bringing about changes in selectivity. In the case of the mobile phase, changing the type and amount of organic solvent, as well as the pH, are all important factors to consider. In many cases, changing the stationary phase chemistry is the most impactful, albeit more expensive, means of changing selectivity. These are all important options to keep in mind when thinking about how to improve a separation.
References
(1) Stoll, D. R. What Are My Options to Improve My Separation? Part 1: Foundational Concepts. LCGC Europe 2023, 36 (4), 132–136.
(2) Snyder, L. R.; Dolan J. W. Introduction. In High-Performance Gradient Elution: The Practical Application of the Linear-Solvent-Strength Model, John Wiley & Sons, 2007; pp. 1–22.
(3) Davis, J. M.; Giddings, J. C. Statistical Theory of Component Overlap in Multicomponent Chromatograms. Anal. Chem. 1983, 55, 418–424. DOI: 10.1021/ac00254a003
(4) Schure, M. R.; Davis, J. M. The Simple Use of Statistical Overlap Theory in Chromatography. LCGC Europe – Recent Developments in UHPLC and HPLC 2015, 33, 8–14.
(5) Snyder, L. R.; Dolan, J. W.; Lommen, D. C. Drylab Computer Simulation for High-Performance Liquid Cromatographic Method Development. J. Chromatogr. A. 1989, 485, 65–89. DOI: 10.1016/S0021-9673(01)89133-0
(6) Dolan, J. W. Back to Basics: The Role of pH in Retention and Selectivity. LCGC Europe 2017, 30, 30–33.
(7) Dolan, J. W.; Snyder, L. R. The Hydrophobic-Subtraction Model for Reversed-Phase Liquid Chromatography: A Reprise. LCGC North Am. 2016, 34, 730–741.
(8) Dolan, J. W.; Stoll, D. R.; Snyder, L. R. How to Take Maximum Advantage of Column-Type Selectivity. LCGC Europe 2017, 30, 484–489.
About the Column Editor

Dwight R. Stoll is the editor of “LC Troubleshooting”. Stoll is a professor and the co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota, USA. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 75 peer-reviewed publications and four book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: amatheson@mjhlifesciences.com

















Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.





Troubleshooting Everywhere! An Assortment of Topics from Pittcon 2025
April 5th 2025In this installment of “LC Troubleshooting,” Dwight Stoll touches on highlights from Pittcon 2025 talks, as well as troubleshooting advice distilled from a lifetime of work in separation science by LCGC Award winner Christopher Pohl.

