Validation, Qualification, or Verification?
LCGC North America
The authors take a look at three recently published documents in the validation literature from groups working to (hopefully!) clear up some of the potential for confusion.
When it was first convened 14 years ago, the International Conference on Harmonization (ICH) ushered in a new era of increased global communication in the regulated pharmaceutical industry that continues to this day (1). But these days it seems that not a month goes by that there is not at least one conference or symposium focusing on one aspect or another of the regulatory landscape. Indeed, a virtual cottage industry has developed specializing in method development and validation. However, like most situations in life, there is good news and bad. The good news: more information containing more details is available now than ever before. The U.S. Food and Drug Administration (FDA) continues to issue new guidances and updates, the United States Pharmacopeia (USP) continues to update its general chapters to further reflect implementation of the ICH guidelines, and industry groups continue to meet with regulatory representatives to help clarify issues. The bad news: all of this new information can lead to confusion and sometimes can be overwhelming. In this month's installment of "Validation Viewpoint," we take a look at three recently published documents in the validation literature from groups working to clear up some of the potential for confusion.



Michael E. Swartz
Some basic definitions — validation, qualification, and verification: In the most general sense, validation refers to a process that consists of at least four distinct components or steps: software, instruments, methods or procedures, and system suitability (2). The system, the software, and the method must all be validated, and system suitability is used to keep the process in check. But while the overall process is called validation, some of the steps also are referred to by that same term, as well as others, such as qualification and verification.


Ira S. Krull
In March 2003, the American Association of Pharmaceutical Chemists (AAPS), the International Pharmaceutical Federation (FIP), and the International Society for Pharmaceutical Engineering (ISPE) cosponsored a workshop titled "A Scientific Approach to Analytical Instrument Validation." Among other objectives, the various parties (the event drew a cross-section of attendees, users, quality assurance specialists, regulatory scientists, consultants, and vendors) agreed that processes are "validated" and instruments are "qualified," finally reserving the term validation for processes that include analytical methods and procedures and software development (3). Use of the term qualification in this sense, however, should not be confused with the same term used to refer to the qualification of impurities in the ICH guideline on impurities in drug substances (4). Qualification in this sense refers to the process of acquiring and evaluating data for the biological safety of impurities.
Verification is a more recent term that refers to the suitability of a compendial procedure under actual conditions of use. The International Organization for Standardization (ISO) puts it this way: "Validation is confirmation, through the provision of objective evidence that the requirements for a specific intended use or application have been fulfilled" (5). "Verification is confirmation, through the provision of objective evidence that specified requirements have been fulfilled" (6). Put simply, if you are using a compendial method, you verify; a noncompendial or alternative analytical procedure, you validate. If this seems confusing, do not worry, more explanation is on the way!


Validation Viewpoint
Method (Procedure) Validation
Earlier this year, the USP proposed revisions to the method validation guidelines published in Chapter 1225 (7). For the most part, the revisions were made to continue to harmonize with ICH terminology, for example, using the word "procedures" instead of "methods." The term "pharmaceutical products" is replaced by the term "pharmaceutical articles" to indicate that the guidelines apply to both drug substances and drug products. A major welcome change is the use of the term "intermediate precision" and the deletion of the section and use of the term "ruggedness," which most people usually confused with robustness anyway. Use of the term ruggedness has been falling out of favor ever since implementation of the original ICH guideline on terminology (8). However, confusion still exists as to exactly what constitutes robustness versus intermediate precision. The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small but deliberate variations in procedural parameters listed in the procedure documentation and provides an indication of its suitability during normal usage. Robustness usually is investigated during the development of a procedure and is useful to establish system suitability parameters. Intermediate precision refers to random events or within-laboratory variations due to such things as different analysts or instruments and results on different days. A rule of thumb: if it is written into the method (for example, 30 °C, 1.0 mL/min, 254 nm), it is a robustness issue. If it is not specified in the method (for example, you would never specify: Steve runs the method on Tuesdays on instrument six), it is an intermediate precision issue. The proposed new list of data elements required for validation is shown in Table I.

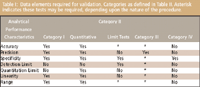
Table I: Data elements required for validation. Categories as defined in Table II. Asterisk indicates these tests may be required, depending upon the nature of the procedure.
Verification of Compendial Procedures
Also earlier this year, the USP published a proposed new chapter, Chapter 1226, entitled: "Verification of Compendial Procedures" (9). The USP says the purpose of this new general information chapter is to provide guidelines for verifying the suitability of a compendial procedure under conditions of actual use. It always has been assumed that USP methods are validated, but not knowing what might have passed for validation when the method was submitted often leads analysts down a path of partial or revalidation, and chapter 1225 does not provide any guidance on how to verify procedures in the absence of a full validation protocol. This new chapter summarizes what is necessary to confirm that the compendial procedure works for a particular drug substance, excipients, or dosage form by verifying a subset of validation characteristics rather than completing a full validation. It is considered an extension of chapter 1225, and both chapters use similar terminology. The intent is to provide guidance on how to verify that a compendial procedure that is being used for the first time will yield acceptable results utilizing the laboratories' personnel, equipment, and reagents. Verification consists of assessing selected "Analytical Performance Characteristics," described in chapter 1225 to generate appropriate relevant data as opposed to repeating the entire validation process.
The Verification Process
The verification process is made up of six components: laboratory personnel, an approved procedure or protocol, data comparison, acceptance criteria evaluation, the final summary documentation, and corrective action, if necessary.
Laboratory personnel must have the appropriate experience, knowledge, and training to be able to carry out the procedure (10). They must be able to accomplish the given functions in the lab, such as operating instrumentation and signing off that analyses were performed as required. It is important to note that it is not enough just to be able to push buttons to make instrumentation function and follow the standard operating procedures (SOPs). GMP requirements put pressure on lab management and personnel to understand the background or basics of any analytical technique that is used in the lab (11,12). But in spite of these requirements, the FDA still frequently cites firms for a lack of trained personnel.
An approved verification document or SOP is needed that describes the procedure to be verified, establishes the number and identity of lots or batches of articles that will be used in the verification, details the analytical performance characteristics to be evaluated, and specifies the range of acceptable results. This verification document should also detail and justify any deviations from the recommendations in chapter 1226. The document should also establish the acceptance criteria that will be used to determine that the compendial procedure performs suitably.
Once samples are analyzed, the data must be scrutinized and compared to the predetermined acceptance criteria in the approved verification document. The final summary documentation should include a summary of the data, the assessment of the results compared to the acceptance criteria, and a decision as to whether or not the data is acceptable, which is a final indication that the laboratory personnel are capable of successfully performing the compendial procedure in the particular laboratory. Acceptable results are final proof that the USP procedure will perform as intended.
If the acceptance criteria are not satisfied, it is necessary to identify the source of the problem, take corrective action, amend the verification document if necessary, and repeat the analysis. The initial unacceptable results, the probable cause, and any corrective actions implemented should also be described in the final summary document.
Of course, there is another possible outcome in which, after several attempts, the verification of the compendial procedure cannot be made. If the source of the problem cannot be identified and rectified, then it can be concluded that the procedure might not be suitable for use with the article being tested. It might then be necessary to revise the current procedure or redevelop and validate an alternative procedure. In any case, the final verification document should summarize the inability to verify the compendial procedure and describe the action taken.

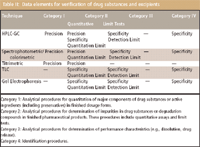
Table II: Data elements for verification of drug substances and excipients
As mentioned previously, Table I lists the analytical performance characteristics that are determined for different categories of assays to ensure validation. But not all of the characteristics listed in Table I need to be repeated for verification of a compendial method; depending upon the type of assay to be verified, different performance characteristics are determined. For analytical techniques applied to drug substances and excipients, the characteristics listed in Table II should be determined. The corresponding characteristics for analytical techniques applied to dosage forms are summarized in Table III.

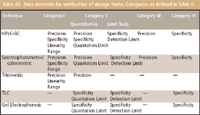
Table III: Data elements for verification of dosage forms. Categories as defined in Table II.
Note that for dosage forms, the number of characteristics to be determined is greater due to the fact that the drug product is usually more complex than either the excipients or drug substance.


Table IV: Summary of precision and system suitability results for an HPLC stability indicating assay with two active ingredients and two degradation products. Triamterine (TMT) and hydrochorthiazide (HCT) are the active ingredients, while 5-nitroso-2,4,6-triaminopyrimidine (NTAP) and 4-amino-6-chloro-1,3-benzenesulfanamide (ACBS) are the related substances called out in the compendial procedure. Average of six replicates at 0.63 mg/mL each. Rt. is retention time, Rs. is resolution, and N is efficiency or plates.
Finally, it is recommended in the guideline that if the procedure will be used by more than one analyst or transferred to another location, intermediate precision should be investigated. (Actually, the document still uses the term ruggedness, and improperly applies the term robustness, something that should be corrected before the guideline is finalized.)


Table V: Determination of the quantitation limit. The quantitation limit was calculated according to established USP and ICH guidelines by the formula: QL = 10STD/S, where STD is the average standard deviation of the response, and S is the slope of the calibration curve (14).
Let us look at an example of a study using a quantitative stability-indicating assay (Category II) run to verify a compendial procedure. In this example, the analysts wished to verify a compendial procedure using a more modern, up-to-date HPLC column. Using Table III, for a quantitative, Category II HPLC assay, precision, specificity, and the quantitation limit must be evaluated. Specificity was evaluated using photodiode-array peak-purity algorithms, which have been covered in a previous installment of "Validation Viewpoint" (13). Table IV summarizes the precision results and Table V the results from the determination quantitation limit. Figure 1 illustrates the actual separation at the quantitation limit used to verify the calculated limit. Peak number one (N-TAP, highlighted in red) is at the calculated (Table V) quantitation limit. Precision data at the quantitation limit also was evaluated (data not shown).

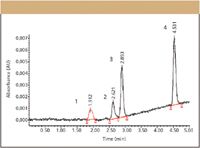
Figure 1: Verification of compendial procedure quantitation limits. Separation was performed on an Alliance 2695 Separations Module (Waters, Milford, Massachusetts). Column: 100 mm X 4.6 mm, 3.5-μm XTerra RPC18; mobile phase A: 10 mM ammonium carbonate, pH 9.0; mobile phase B: methanol; gradient: 15â90% B (linear) over 5 min; flow rate: 1.0 mL/min; temperature: 34 °C; injection volume: 20 μL; detection: UV absorbance at 280 nm. Peaks: 1 = NTAP (highlighted in red), 2 = ACBS, 3 = HCT, 4 = TMT.
Analytical Instrument Qualification
As mentioned earlier, processes are "validated" and instruments are "qualified." Analytical instrument qualification (AIQ) provides documented evidence that the instrument performs suitably for its intended purpose and that it is properly maintained and calibrated. Qualification normally is grouped into four distinct phases, design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ). A definition and discussion of each of the qualification phases have been covered in previous "Validation Viewpoint" columns and elsewhere and really do not need to be repeated here (2,14). The AAPS publication also does an excellent job of capturing the definition and documentation of each of these phases as well, as an excellent discussion on software validation to which the reader is referred for more information (3). However, there are a couple of other significant areas that the AAPS group focused on that are worth noting; documentation and the categorization of instruments according to the level of qualification required.
Both static and dynamic documentation can result from an AIQ. Static documents are generated during the DQ, IQ, and OQ phases and should be kept in a separate qualification binder. Static documents can include such things as user manuals, site requirement documents, etc. Dynamic documents are generated during the OQ and PQ phases, when actual instrument testing takes place. These documents provide a running record for the instrument use and maintenance and should be kept in a system log book with the instrument, available for viewing as necessary by anyone interested (that is, the FDA). These documents also should be appropriately archived for future reference and protection.
Instruments were placed into three categories (A, B, and C), again based upon their complexity and proposed level of qualification.
The conformance of Group A instruments to user requirements is determined by visual observation; no independent qualification process is required. Examples of Group A instruments include spatulas, ovens, magnetic stirrers, microscopes, and vortex mixers.
The conformance of Group B instruments to user requirements is determined according to the instruments' SOP, and their failure usually is readily discernable. Examples of instruments that fall into this category are pH meters, balances, thermometers, refrigerator–freezers, and vacuum ovens.
Group C instruments are defined as highly method-specific, complex instruments with conformance determined by their application. Full qualification as outlined in the AAPS report is applied to instruments in this group. Examples include high performance liquid chromatography (HPLC) and gas chromatography (GC) instruments, spectrometers, mass spectrometers, and electron microscopes.
Conclusion
Data quality is built on the foundation of procedure validation and verification, software validation, AIQ, and system suitability. Each of these components plays a critical role in the process of validation. The three documents highlighted in this column (3,7,9) will go a long way toward clearing up potential avenues of confusion in the industry and certainly should be consulted for additional details beyond those that we can cover in this short column. It should be noted, however, that none of these three documents has been finalized in any way, but are merely proposals before their various organizations, and we shall certainly keep you informed of any developments in subsequent columns. Hopefully we'll also be able to report that all of the confusion surrounding robustness and ruggedness is finally put to rest once and for all!
Acknowledgments
The authors would like to acknowledge Michael D. Jones of Waters Corporation, Milford, Massachusetts, Jerry Lanese of the Lanese Group, Leawood, Kansas, and Paul Newton of GlaxoSmithKline, RTP, North Carolina, for contributions to this manuscript.
Michael E. Swartz "Validation Viewpoint" Co-Editor Michael E. Swartz is a Principal Scientist at Waters Corp., Milford, Massachusetts, and a member of LCGC's editorial advisory board.
Ira S. Krull "Validation Viewpoint" Co-Editor Ira S. Krull is an Associate Professor of chemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board..
The columnists regret that time constraints prevent them from responding to individual reader queries. However, readers are welcome to submit specific questions and problems, which the columnists may address in future columns. Direct correspondence about this column to "Validation Viewpoint," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com
References
(1) www.ich.org.
(2) M.E. Swartz and I.S. Krull, Analytical Method Development and Validation (Marcel Dekker, New York, 1997).
(3) AAPS PharmSciTech 2004, 5(1) Article 22 (http://www.aapspharmscitech.org).
(4) ICH Q3A(R), "Impurities in New Drug Substances," Federal Register 68(68), 6924–6925. See also: www.ich.org.
(5) ISO 9000:2000 clause 3.8.5.
(6) ISO 9000:2000 clause 3.8.4.
(7) Pharmacopeial Forum 31(2), 549 (Mar./Apr. 2005).
(8) ICH Q2A, Federal Register 60, p. 11260. See also www.ich.org.
(9) Pharmacopeial Forum 31(2), 555 (Mar./Apr. 2005).
(10) M.E. Swartz, I.S. Krull, and J. McCabe, LCGC 22(9), 906 (2004).
(11) Current Good Manufacturing Practice for the Manufacture, Processing, Packing, or Holding of a Drug Product, 21 Code of Federal Register (CFR) Part 211; Subpart A: General Provisions 211.1-Scope.
(12) Current Good Manufacturing Practice for the Manufacture, Processing, Packing, or Holding of a drug Product , 21 Code of Federal Register (CFR) Part 211; Subpart B: Organization and Personnel 211.25-Personnel Qualifications.
(13) M.E. Swartz and I.S. Krull, LCGC 23(6), 47 (2005).
(14) M.E. Swartz and I.S. Krull, LCGC 16(10), 922 (1998).













Analyzing Vitamin K1 Levels in Vegetables Eaten by Warfarin Patients Using HPLC UV–vis
April 9th 2025Research conducted by the Universitas Padjadjaran (Sumedang, Indonesia) focused on the measurement of vitamin K1 in various vegetables (specifically lettuce, cabbage, napa cabbage, and spinach) that were ingested by patients using warfarin. High performance liquid chromatography (HPLC) equipped with an ultraviolet detector set at 245 nm was used as the analytical technique.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

The Effect of Time and Tide On PFAS Concentrations in Estuaries
April 8th 2025Oliver Jones and Navneet Singh from RMIT University, Melbourne, Australia discuss a recent study they conducted to investigate the relationship between tidal cycles and PFAS concentrations in estuarine systems, and offer practical advice on the sample preparation and LC–MS/MS techniques they used to achieve the best results.

Assessing Safety of Medications During Pregnancy and Breastfeeding with LC–MS/MS
Published: April 8th 2025 | Updated: April 8th 2025Researchers conducted a study on medications used in psychiatry and neurology during the perinatal period, assessing how antiepileptic drugs (AEDs) affect placental functions, including transport mechanisms, nutrient transport, and trophoblast differentiation. Several quantitative methods, such as those for antianxiety and hypnotic drugs, were established to evaluate the safety of pharmacotherapy during breastfeeding using liquid chromatography-tandem mass spectrometry (LC–MS/MS).

