Using Two-dimensional Liquid Chromatography to Separate Complex Mixtures of Peptides
LCGC Europe
Two-dimensional liquid chromatography (2D-LC) allows analysts to deal with complex samples that either cannot be adequately separated by one-dimensional liquid chromatography (1D-LC) or require excessively long analysis times. Peptide mixtures, whose characterization is relevant in many areas (e.g., proteomics, food analysis, pharmaceutical, life sciences), are a clear example of such complexity. An overview of the most used 2D-LC modes of operation is presented and several examples of their use for the separation of peptide mixtures are described.
The characterization of complex mixtures of peptides is relevant in numerous applications, including proteomics, food analysis, pharmaceutical and life sciences. One-dimensional liquid chromatography (1D-LC) cannot deal with the complexity of these peptide mixtures, not even if the most efficient columns are used in combination with the most advanced mass spectrometric detection devices. The resolution is simply insufficient. To overcome this limitation, multidimensional separation systems, where the dimensions are based on different separation mechanisms, have been introduced.1 In this article we will assess the potential of using two-dimensional liquid chromatography (2D-LC) to separate peptide samples. Several routes aimed at improving resolution in peptide analysis are discussed from a practical perspective and different instruments used for this purpose are compared.
Peak Capacity in LC
The ability of a chromatographic system to deal with complex samples depends on the number of peaks the system can separate relative to the number of compounds present. A useful metric here is the peak capacity. Peak capacity is defined as the maximum number of peaks that can be separated within a given retention window at a desired degree of resolution.2 When peptide analysis is concerned, peak capacities obtainable in 1D-LC separations typically range from 100–400.3 As peak capacity generated by 1D-LC is often not enough to deal with the complexity of peptide mixtures from, for example, protein digests, 2D-LC has been applied with the aim of improving the separation power and reducing sample complexity to an acceptable level prior to tandem mass spectrometry (MS–MS) analysis.4
2D-LC consists in subjecting the whole sample, or just the part of interest, to two distinct and consecutive separations. If the whole sample is subjected to the 2D analysis, the system is said to be comprehensive and it is indicated as LC×LC;5 if just a fraction is transferred the term heart-cutting 2D-LC is used. In both set-ups two separation systems — the first dimension (1D) and the second dimension (2D) ones — showing enough difference in selectivity are employed. The main appeal of LC×LC lies in the fact that, if the separation mechanisms in the two dimensions are completely independent and separation from the 1D is maintained, the total peak capacity amounts to the product (and not the sum!) of the peak capacity in the two dimensions.6 LC×LC is described in more detail below.
The Different Approaches to LC×LC
In an articulate and comprehensive communication, Guiochon et al. have recently reviewed the most important achievements in the field of LC×LC obtained so far.7 Their classification of the different LC×LC methods is summarized below.
LC×LC can be performed in two different ways: so called off-line operation requires the eluate from the 1D to be collected as narrow fractions and stored. Fractions of interest are, subsequently, reinjected onto the 2D column. On-line LC×LC analysis, on the contrary, requires an automated set-up. Fractions from the 1D analysis are automatically transferred to the 2D column. Here a rapid separation is performed allowing the 2D column to receive the next fraction and be ready for a new separation.
Off-line implementation has both advantages and drawbacks over on-line operation. The lack of a time constraint for the 2D analysis allows the use of columns that are longer and more efficient than those typically used in on-line LC×LC. As a result, a very high resolving power can be obtained. If that is done, however, the technique becomes rather slow. The second practical advantage of the off-line approach is the extreme flexibility in the operation: neither the frequency of fraction collection nor that of the analyses performed in the 2D need to be constant, let alone equal. It is not even required to analyse all the collected fractions or to run them in the order in which they were collected. The corresponding drawback here is the management of the large number of collected fractions: care in storing them is required, complicating the experimental set-up. Fraction storage offers the possibility to concentrate the fractions prior to injection. If this is done, great care must be taken to avoid phenomena that could pollute them.
When using on-line LC×LC operation instead of the off-line route, the chance for the operator of "spoiling" the sample is minimized. Another important advantage here is that the 1D separation is maintained. In contrast, an important disadvantage is that the setting up of an automated on-line system is time consuming because many parameters for each of the two dimensions (column dimensions, flow-rate, total pressure, optimum sampling period, etc.) need to be taken into account and optimized. Moreover, as already mentioned, the separation in the 2D suffers from a serious time constraint: a run has to be completed before the next fraction can be transferred. Therefore, rather short columns have to be used in the 2D, the exact length and analysis time allowed depending on the speed or peak width in the 1D.
In comprehensive on-line 2D analysis, the 1Dcolumn can be operated in two different ways: the most common and widespread used one is continuous low-flow operation;8 the other option is that of alternating high flow and zero flow operation, that is, using stop-flow.9 Below, a comparison between these two on-line modes will be given, focusing particularly on the different instruments and the pros and cons of the two modes.
Column choice in LC×LC of Peptides
Because of its very high efficiency and selectivity, reversed-phase (RP) LC–MS is by far the most popular technique when the analysis of peptides is concerned. In case the RPLC separation results in a too high number of coeluting peaks for MS to resolve, one further dimension can be added to the system resulting in an LC×LC method. Several combinations of separation modes have been used for that purpose, the most popular ones being:
SCX–LCRPLC: Strong cation-exchange (SCX) beds, separating compounds mainly according to their charge, have been used as the 1D in LC×LC systems by several research groups. This combination of separation phases has been exploited both in on-line10 and off-line operation.11
SECXRPLC: Size exclusion chromatography (SEC), separating analytes according to size, has also been often used as the 1D in LC×LC systems. This column combination has been exploited by Opiteck et al.12 for continuous low-flow LC×LC and by Bedani et al. for setting up instrumentation operated in the stop-flow mode.9
RPLCXPLC: A more and more common approach in peptide analysis is to exploit the separation power of two RPLC beds showing different selectivity and/or "forcing" different selectivity by changing parameters such as pH, temperature or modifier type.13
Instrument Design and Interface for LC LC of Peptides
Off-line LC×LC operation requires the same components — pumps, injector, column(s), detector(s) etc. — as 1D-LC, needing one more column for the 2D. This is true for most applications, although it can be argued that LC×LC is also possible if the mobile phase — and not the stationary phase — is changed in the two dimensions.13 After collecting fractions, the 1D column is replaced by the 2D one and the fractions of interest are used as the new samples and reinjected. A fraction collector can be one useful "extra" to the instrumentation, resulting in less critical operator involvement.


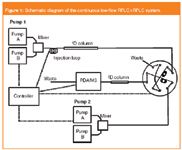
Figure 1
On-line operation requires automated instrumentation. As a consequence (at least) one more LC pump than for 1D systems is needed so that the first and second dimension columns can be run at the same time. On the contrary, no further detector devices than for 1D-LC instrumentation are in principle necessary, if detection is achieved just after the 2D separation. In Figure 1 the schematic of a typical instrument for on-line continuous low-flow operation is shown.14 This instrumentation uses a two-position ten-port valve as the interface between the first and second dimension columns (see Figure 2) as described by van der Horst et al.:8 fractions of the effluent from the 1D column are alternatingly collected in two loops. When the valve is switched, the stored fraction is injected onto a second column, where they get separated further. The system in Figure 1 refers to an application requiring gradients in both dimensions: two binary pumps can be used for that purpose.

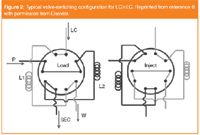
Figure 2
In Figure 3, a schematic of the instrumentation for stop-flow analysis is presented. The 1D outlet is connected to a six-port valve: in the "transfer"-position the effluent from the 1D is directed to the 2D and the 2D pump is connected to a flow restrictor, which keeps the pressure constant during the actual transfer; in the "analysis"-position, the 1D pump is connected to a stop-flow valve and the 2D one to the inlet of the 2D column. Before the first peak elutes the stop-flow valve is open, letting the hold-up volume go to waste. When the transfer of the first fraction starts, the stop-flow valve is switched to the "stop-flow"-position. Because of that, when switching the 1D pump off, the pressure in the 1D stays constant. At the end of the analysis the stop-flow valve is switched back to the initial "waste"-position.

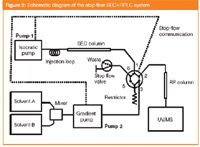
Figure 3
When instrument design is concerned, stop-flow operation offers some advantages over the continuous low-flow method. In an on-line 2D analysis part of the separation from the 1D is lost as a result of diffusion. To minimize this loss, the transfer of the collected fractions from the end of the 1D column to the head of the 2D column should occur with a minimum of additional band dispersion and, for that reason, the volume between the first and the second dimension should be as little as possible. When using stop-flow operation this problem is less serious: as already mentioned, the interface is now reduced to just one two-position six-port valve. No storage loops are present as the fractions are transferred straight away from the first to the second dimension column. For peptide analysis this is even more appealing. It is, in fact, well-known that two factors are complicating the development of on-line systems for peptide analysis:
1. Non-polar peptides are very susceptible to adsorption in all types of interfaces such as storage loops, tubing etc.
2. Peptides are very prone to give time shifts depending on column quality and exact gradient composition.
Examples of 2D-LC Separations of Peptides
Numerous interesting applications for 2D-LC of peptides have been published in the last few years, both in the off- and on-line mode.9–12,15 Recently, Marchetti et al.11 published an off-line separation of the tryptic digest of bovine serum albumin (BSA) using a 1D 4.6 mm × 110 mm SCX column (5 μm particle size) with a peak capacity of 54 and a 2D 4.6 mm × 50 mm column packed with porous shell particles (average diameter 2.7 μm, shell thickness 0.5 μm) having a peak capacity of 140; 182 fractions from the 1D were analysed in a total analysis time of 27 hours with an overall peak capacity estimated at about 7000. Clearly, time is the currency in which we have to pay for separation power.
In on-line LC×LC, the current trend is to perform very fast 2D separations. This trend also applies to the analysis of complex mixtures of peptides. The result is an important gain in separation power for a negligible increase in the analysis time compared with 1D-LC operation. To achieve that, the two most common strategies are to use in the 2D either monolithic columns or high-temperature HPLC.10,15 Monolithic columns are about one order of magnitude more permeable than packed columns of comparable efficiency: it is thus easy to operate them much faster and also to generate faster the required peak capacity. Kimura et al.15 used a short (4.6 mm × 25 mm) monolithic silica C18 column as 2D column in combination with a 1D ion exchange (2.1 mm × 50 mm) column packed with 5 μm polymer-based SCX beads for the separation of tryptic digests of BSA. Two minute fractionation in the 1D allowed one minute for gradient separation in the 2D, resulting in a peak capacity of about 700 in 40 min. The second approach to speed-up elution is the use of high temperatures. When columns are operated at higher temperature, their efficiency remains nearly constant while the value of the optimum linear velocity increases. As a result, the column can be operated at higher mobile phase velocity resulting in a decrease of the total analysis time. Stoll et al.10 separated a tryptic digest of BSA using a 1D 2.1 mm × 50 mm SCX column packed with 5 μm particles and an RPLC column carefully chosen for its stability in hot, acidic mobile phases (SB-C18, 3.5 μm, 2.1 mm × 50 mm) in the 2D. Under typical conditions they were able to reduce the effective gradient time to just 16 s, for a total cycle time of 21 s. A complete 20 min LC×LC analysis provided a peak capacity of about 1350.
Below, a few examples covering both off-line and on-line approaches are presented. All chromatograms shown refer to separations of Biozate3, a whey protein hydrolysate. For all systems discussed, mobile phase A was water containing 0.1% trifluoroacetic acid (TFA) and B was acetonitrile (AcN) also containing 0.1% TFA.
The off-line approach in Figure 4 shows a separation that is obtained when using, for both dimensions, RP columns with different selectivity: the sample is run on a 1D 4.6 mm × 250 mm C18 column packed with 5 μm particles (see Figure 4, left-hand side), 20 s wide fractions are collected and reinjected on a 2D 4.6mm × 250 mm alkylamide bed also packed with 5 μm particles. The same gradient was used in the two dimensions: it started at 5% B and ended at 50% B in 30 min; at 35 min it would increase to 65% B and go back to 5% B at 36 min. As it is clear from Figure 4 (see right-hand side), a typical 2D separation increases the peak capacity in the 1D by approximately a factor 10 (see caption of Figure 4 for experimental conditions).

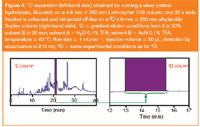
Figure 4
The chromatogram in Figure 5 was acquired using the system shown in Figure 1. RPLC columns were used in both dimensions:14 the 1D used a 4.6 mm × 250 mm Luna CN column packed with 5 μm particles; the 2D consisted of a 4.6 mm 50 mm Inertsil ODS-3 C18 column also packed with 5 μm particles. A typical 1D gradient started at 5% B and ended at 23.5% B either in 2 or 4 h. The separation in the 1D was run at 0.050 or 0.100 mLmin–1. For the 2D, gradients were optimized for each effluent slice to be injected in the 2D. Differently from what already reported in literature,12 it was here decided not to run two 2D columns in parallel although this would translate in an undoubted decrease in total analysis time: the strong dependence of peptide separation on column quality and exact gradient composition is likely to result in fluctuations in retention times in the 2D. Because of that, the use of such a 2D design does not appear so appealing.
The chromatogram in Figure 6 refers to a separation obtained when running SEC PLC instrumentation using stop-flow operation (see Figure 3).9 The 1D was a 7.8 mm × 300 mm TSK-Gel G2500PWXL SEC column (6 μm particles) with a 0–3000 Da range, eluted with water containing 0.1% TFA (A) at 0.37 or 0.50 mLmin–1 . The 2D column was a C18 silica monolith 4.6 mm × 25 mm Chromolith Flash column extended with a 10 mm long Chromolith guard column, eluted with a gradient of A and AcN with 0.1% TFA (B). A 30 s effluent slice was transferred from the first to the second dimension. At the end of the 30 s, the six-port valve was switched and the gradient started: the flow-rate would increase to 2 mLmin–1 in the next 30 s and, after that, the solvent programming would start. The practical reason for the gradual flow increase was to avoid pressure shocks during the actual transfer (the maximum pressure tolerable by the 1D column was only 43 bars). A typical gradient started at 5% B; at 2.50 min B was 40% and, at 3.50 min, 90%. B was then maintained at 90% up to 5.50 min and, at 6.50 min, was decreased to 5%. The total cycle was 10 min and elution of the first column was resumed before the next cycle. The useful duration of the cycle was 5 min, generating a 2D peak capacity of 30. The total peak capacity obtained from the SEC PLC system was 300, being the product of the 1D peak capacity (30) and that of the 2D column (10). Unlike continuous low-flow operation, when using a stop-flow method the two separations are disconnected and the 2D analyses could, in principle, last as long as needed. For practical reasons, however, this time should not be too high, avoiding having to "park" peaks in the 1D for extremely long times. Contrary to what is commonly thought, stop-flow operation does not result in additional band broadening in respect with a continuous low-flow one;9 nonetheless, when total analysis time is too high, band broadening, as a result of longitudinal diffusion, becomes serious for late-eluting compounds.


Figure 5
Conclusions
Off-line coupling is more flexible than on-line operation regarding the choice of the 2D column, as no 2D time constraint is required. Off-line operation is the most powerful approach to LC×LC in terms of resolution but, unfortunately, is also the slowest one. On-line operation permits a marked decrease of the total analysis time and makes the handling of intermediate fractions easier than off-line coupling. It can be performed using continuous low-flow or stop-flow.


Figure 6
RP-LC is the most used technique for peptide separation and has often been used as the 2D in LC×LC, allowing a straightforward coupling to MS. The particular "nature" of peptides favours the use of LC×LC instruments which minimize interfaces; the strong dependence of peptide retention times on column quality and exact gradient composition makes the use of two 2D columns in parallel difficult. Stop-flow operation is probably, therefore, more attractive to use.
Acknowledgments
Laurie Davis from Davisco Foods International Inc. (Eden Prairie, Minnesota, USA) is kindly acknowledged for donating Biozate3.
Filippo Bedani is a PhD student at the University of Amsterdam. During his PhD he mostly focused on improving the resolution power of on-line two-dimensional liquid chromatography systems for the analysis of biomacromolecules.
Hans-Gerd Janssen is group-leader chromatography and mass spectrometry at Unilever R&D, Vlaardingen, the Netherlands. He also holds a part-time chair in biomacromolecular analysis at the Van 't Hoff Institute for Molecular Sciences of the University of Amsterdam, Amsterdam, the Netherlands.
References
1. F. Erni and R.W. Frei, J. Chromatogr. , 149, 561–569 (1978).
2. J.C. Giddings, Anal. Chem. , 39, 1027–1028 (1967).
3. M. Gilar et al., J. Chromatogr. A , 1061, 183–192 (2004).
4. T. Wehr, LCGC N. Am. , 20, 954–962 (2002).
5. J. Blomberg et al., J. High. Resolut. Chromatogr. , 20, 539–544 (1997).
6. B.L. Karger, L.R. Snyder and C. Horvath, An Introduction to Separation Science ; Wiley and Sons, New York, (1973).
7. G. Guiochon et al., J. Chromatogr. A , 1189, 109–168 (2008).
8. A. van der Horst and P.J. Schoenmakers, J. Chromatogr. A , 1000, 693–709 (2003).
9. F. Bedani et al., J. Chromatogr. A , 1133, 126–134 (2006).
10. D.R. Stoll and P.W. Carr, J. Am. Chem. Soc. , 127, 5034–5035 (2005).
11. N. Marchetti, J.N. Fairchild and G. Guiochon, Anal. Chem. , 80, 2756–2767 (2008).
12. G.J. Opiteck, J.W. Jorgenson and R.J. Anderegg, Anal. Chem. , 69, 2283–2291 (1997).
13. M. Gilar et al., Anal. Chem. , 77, 6426–6434 (2005).
14. F. Bedani, W.Th. Kok and H.-G. Janssen, manuscript submitted for publication.
15. H. Kimura et al., J. Sep. Sci. , 27, 897–904 (2004).















Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



