The Use of Thin-Layer Chromatography with Direct Bioautography for Antimicrobial Analysis
This article gives an overview of screening methods for the detection of antimicrobials, including dilution and diffusion methods and bioautography, mainly direct bioautography. The thin-layer chromatography method with direct bioautography was worked out to analyse enrofloxacin and ciprofloxacin residues in milk.
Bioautography is a laboratory technique to detect substances affecting the growth rates of test organisms in complex mixtures and matrices. The method is based on the biological activity of the analyte, which can be antibacterial, antifungal, antitumour, antiprotozoae etc. The main fields of bioautography are
- searching for new antibiotic substances and new antifungal, antitumour and antiprotozoae compounds by studying biological activities of substances originating from plants, microorganisms or combinatorial chemistry
- investigation of antibiotics and other biologically-active compounds in waste water, drinking water, body fluids, feed and food
- quality control of antibiotic pharmaceuticals
- searching for antimicrobial compounds effective against plant pathogenic bacteria and fungi
- detection and determination of toxic (e.g., aflatoxins) or phototoxic (e.g., furocoumarins) compounds.



Bioautography belongs to a large group of screening methods for the detection of antimicrobial activity. Diffusion and dilution methods are also used.
Diffusion Methods
Diffusion methods include agar-overlay techniques such as disk, strip, well (hole) and cylinder. The disk diffusion assay is one of the most commonly used methods of antimicrobial susceptibility testing.1–5 In the USA this is the official method for qualitative detection of inhibitory substances in milk.2 Small filter paper disks (about 1 cm diameter) impregnated with a standard amount of antibacterial substances are placed onto a previously inoculated agar plate for this method. The plate is inverted and incubated. Sometimes, before incubation, plates with disks are left at temperatures close to 0 ÞC for a few hours to enable diffusion. The incubation time varies from several to 48 hours. The diameter of the zones of bacterial growth inhibition is a measure of susceptibility. The similar method, called E-test, uses strips instead of disks.3 Standard disks and strips containing different antibiotics and different amounts are commercially available.
The official method for quantitative detection of β-lactam residues is the cylinder plate method.4,6 Stainless steel cylinders, approximately 1 cm long, are placed on the inoculated agar surface of a prepared Petri plate. The cylinders are filled with samples and the standards and plates are incubated overnight. Next, plates are inverted to remove cylinders and zones of inhibition are measured.
In the agar well diffusion assay a few millimetre-diameter holes are cut into the inoculated agar surface and the wells are filled with tested antibacterials.5 The plates are left for a few hours at room temperature to allow diffusion into the agar medium, then the plates are incubated and the zones of inhibition are measured. Diffusion methods are usually used for pure substances.
Dilution Methods
Dilution methods are mainly used to determine the minimum inhibitory concentration (MIC) of pure substances and extracts. The sample should be homogeneously dispersed in water. It is usually mixed in various dilutions with inoculated medium. After incubation, inhibitory properties of the sample can be estimated by turbidimetric or visual comparison with a control culture. In the tube assay various concentrations of the analyte are mixed in series of tubes with bacteria suspension.7 The smallest amount causing inhibition of bacterial growth — the medium remains clear — gives the MIC value. In agar dilution various concentrations of antibacterial substance are mixed with nutrient agar.3,8 Agar plates are inoculated and incubated. The lowest concentration of antibiotic resulting in no growth is read as the MIC value. The broth microdilution assay is performed in the microtitre plate. After incubation the bacterial growth is indicated by the presence of a white "pellet" on the well bottom.5
Bioautography
Paper chromatography followed by bioautography was used for the first time in 1946 by Goodal and Levi to estimate the purity of penicillin.9 This was called contact bioautography (i.e., the developed paper chromatogram was placed onto the inoculated agar layer enabling the diffusion of antibiotics from paper to agar). Thin-layer chromatography–bioautography was introduced by Fisher and Lautner10 and Nicolaus11 et al. in 1961. The first review covering both paper and thin-layer chromatography–bioautography was written by Betina12 in 1973. The main benefit of bioautography is that it provides information about antimicrobial activities of substances separated from a mixture.
Bioautography methods are usually divided into three categories:
- Agar diffusion or contact bioautography.
- Immersion or agar-overlay bioautography.
- Direct bioautography.
Contact Bioautography
In contact bioautography antimicrobials diffuse from a TLC plate or paper to an inoculated agar plate.13–15 The chromatogram is placed face down onto the inoculated agar layer and left for some minutes or hours to enable diffusion. Then the chromatogram is removed and the agar layer is incubated. The inhibition zones are observed on the agar surface in the places where the spots of antimicrobials are stuck to the agar. The method resembles a disk assay. The disadvantages of contact bioautography were difficulties in obtaining complete contact between the agar and the plate and adherence of the adsorbent to the agar surface. These shortcomings were avoided by applying for chromatography silicic acid-glass fibre sheets, ChromAR.15 Still, the basis of the method was the same and antimicrobials had to be transferred from the sheet to agar causing their loss and dilution.
Immersion Bioautography
In immersion bioautography the chromatogram is covered with a molten, seeded agar medium. After solidification, incubation and staining (usually with tetrazolium dye) the inhibition or growth bands are visualized.11,16–19 Sometimes, before incubation, plates are left for several hours at low temperature to enable diffusion.18 Agar-overlay is a hybrid of contact and direct bioautography. Antimicrobials are transferred from the TLC plate to the agar layer as in the contact assay but during incubation and visualization the agar layer stays onto the plate as in direct bioautography. The main disadvantage of this method is lower sensitivity caused by dilution of antibacterials in the agar layer compared with direct bioautography. Agar overlay is advised especially when direct bioautography is impossible to perform.20
In the 1960s the contact and immersion bioautography in various versions were routinely used. They are described in detail by Betina.12 Nowadays direct bioautography prevails over them.
Direct Bioautography
In direct bioautography a developed plate is dipped in the suspension of microorganisms growing in a suitable broth or this suspension is sprayed onto the plate.21–24 The plate is incubated and microorganisms grow directly on it. Hence, separation, preconditioning, incubation and visualization are performed directly on the plate. For location and visualization of antibacterials tetrazolium salts are usually used, which are converted by the dehydrogenases of living microorganisms to intensely coloured, formazan. The bacteria are killed by antimicrobials on the TLC plate so colour is not produced in the places of antibacterial spots and so-called zones of inhibition that are pale on a coloured background are formed. The most frequently used tetrazolium dye is 3-{4,5-dimethylthiazol-2-yl}-2,5-diphenyltetrazolium bromide,
so-called MTT. Reduction of tetrazolium salts can be increased many times by addition of detergent (e.g., Triton X-100) to the dye. A direct bioassay can also be performed with the luminescent bacteria, such as Photobacterium phosphoreum25 or Vibrio fischeri.26 The plates were dipped fast in a suspension of bacteria, covered with glass plate and bioluminescence was measured by a Peltier — cooled charged coupled device (CCD)25 camera or luminograph video imaging system.26 Alternatively, a photographic procedure with the X-ray film was used.26 The bioluminescence decreases for bacteria exposed to toxic substances as a signal of disturbing vital cell processes. Though there are no changes in bacterial growth, the method can be considered as a direct bioautography.
Direct bioautography is usually performed without agar gel. The plate is dipped in or sprayed with the seeded culture broth of a proper viscosity, which guarantees conditions for the life of test bacteria. Recently, a bioautographic method was developed called "agar spray assay,"27 which can be considered as an "agar version" of direct bioautography. The method was based on the earlier published agar overlay method.28 Dehydrated potato dextrose agar (PDA), cooled after sterilization to 48 ÞC was used to prepare a solution of spores to be sprayed on the TLC plates. Spraying with a manual sprayer was performed rapidly to avoid immediate solidification. The inoculum was solidified at approximately 0.5 mm layer and the plates were incubated at 25 ÞC for at least 60 h in a humid atmosphere until spores produced a dark mycelial mat. Inhibition zones were visible without the need for staining. The similar method was used also by Mackeen et al.,29 in searching for antifungal properties of Garcinia atroviridis, a fruit tree from Malaysia.
Recently, a complex bioautographic system (BioArena™) was developed, which is suitable for the study of antibiotics and toxins.30,31 The system integrates the up-to-date methodological and biological results of direct bioautography with OPLC (overpressured layer chromatography). The attractive solutions of BioArena™ include an optimum, changeable incubation time and the parallel or consecutive application of necrotrophic and/or biotrophic bacteria or fungi. It can be used for the studies of promoting or retarding cell proliferation processes. OPLC-direct bioautography was also used for the detection of aflatoxins caused by antibacterial activity of formaldehyde formed by the bacterial cells from the methoxy groups of aflatoxins molecules. Model interactions between formaldehyde and endogenous substances (L-arginine and glutathione) were proposed.32 It was proved using the BioArena™ system that these endogenous substances influence antimicrobial activity not only of aflatoxins, but also of trans-resveratrol.33 The BioArena™ system is especially suitable for investigating biochemical interactions in an adsorbent bed after the chromatographic separation.
Merck has recently introduced a ready-to-use test for direct bioautography, called Chrom Biodip® Antibiotics.34–36 The kit contains ampoules with Bacillus subtilis spore suspension, bottles with MTT reagents and with nutrient medium. The optimized test culture medium for this purpose consists of peptone, casein, starch and a phosphate buffer. To obtain proper viscosity it also contains 1.5% of bacterial polysaccharide as a gelatinizing agent. The developed TLC plates should be dipped quickly in the inoculated broth and incubated overnight in a humid atmosphere (19 h at 23–35ÞC ). The test can be performed in both microbiological and chemical laboratories.
TLC–direct chromatography can be combined with other chromatographic techniques. Horváth et al. determined the composition of various thyme chemotypes by gas chromatography. Then, their bioactivity against Gram negative plant pathogenic bacteria was examined by direct bioautography.24 Islam et al.27 separated the extract from plant material by column chromatography and examined the obtained fractions by the bioautographic agar spray assay. The direct bioautography was used in the search for new antifungal agents of plant origin in combination with liquid chromatography hyphenated techniques.37 The crude extract was analysed by LC–NMR, LC–MS–MS and LC–UV. The fractions collected from LC microfractionation, following the LCNMR analysis, were used for TLC bioautography. The assay pointed to the LC peaks of antifungal activity. The role of TLC-bioautography as a complementary method to hyphenated techniques in screening bioactive compounds from medicinal plants was discussed by Hostettman et al.38
Rios et al. discuss the factors influencing bioautography results and concluded that because of their diversity and variability, it is difficult to standardize the bioautographic methods.39 Still, the attempts to standardize these parameters or at least to estimate their influence on the final results were made. The factors influencing direct bioautography are exhaustively discussed in the chapter by Botz, Nagy and Kocsis.36 The following parameters are taken into consideration:
- Mobile phases and their additives.
- Type of adsorbent.
- Test microorganism.
- Living conditions for test bacteria (e.g., type of broth, density of inoculum).
- Precondition of plates, mode of development, drying, post-chromatographic conditioning.
- Post-chromatographic detection (e.g., humidity of the chamber, time of incubation, bacteria concentration, detecting reagent).
Optimal life conditions of Gram-positive (Bacillus subtilis) and Gram-negative bacteria (Escherichia coli) were studied by Nagy et al.40–42 The viability of the bacteria on the TLC plates was measured on the basis of their ATP content, which was determined by the bioluminescence luciferin/luciferase assay. The authors found it essential to optimize an optical density of dipping bacterial suspension, the immersion time and number of plates that can be dipped in the same suspension as well as the incubation time. They proposed using bacteria in the exponential phase and a much shorter incubation time (4–8 h for Bacillus subtilis and 3 h for Escherichia coli) compared with that proposed before.34,35 With shorter incubation time other adsorbents than silica could be used (alumina, cellulose or polyamide).
In our laboratory conditions of TLC-direct bioautography of residues of doxycycline and flumequine in milk were established.43,44 The sample pretreatment, development and microbial detection were performed directly on the plate. Spiked milk samples were applied onto the concentrating zone of the TLC plate. The plate was predeveloped with hexane, followed by acetone to remove milk lipids and the proper mobile phase was then used. Milk proteins remained on the surface of concentrating zone while the antibiotics, separated in this way from the milk matrix, migrated freely. The developed plates were subjected to bioautography using the Merck Biodip® test. Next, TLC-direct bioautography of enrofloxacin and ciprofloxacin standards were established.45 It was shown that the size of inhibition zone depends on applied volume of the antibiotic solution. The minimum detectable dose for the standards of the antibiotics was found to be 0.5 ng per spot while the limit of detection was at the level of 0.01 ppm for 50 µL of antibiotic standard solutions applied onto the plate.
The quantitative bioautographic analysis is usually done by the regression analysis of the inhibition zone sizes. According to some papers, the relationship between the diameter or the area of inhibition zone plotted against the logarithm of the concentration of the antimicrobial applied is linear.12,36,46,47 Our experiments show that this approximation is possible only for a narrow range of concentrations (one or two orders of magnitude). For a wider range of concentrations exponential relation fits better. The dependencies between the areas of inhibition zones and the amounts applied of enrofloxacin and ciprofloxacin are plotted in Figure 1. The exponential curves were obtained with a higher correlation coefficient (R2 = 0.9633 for ciprofloxacin, R2 = 0.9524 for enrofloxacin) than the linear curves (R2 = 0.8302 for ciprofloxacin, R2 = 0.8713 for enrofloxacin).

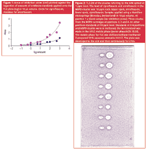
Figure 1 shows it is impossible to establish detection limits from the linear calibration curve in the semi-log scale on the basis of inhibition zone areas obtained for the higher range of standards. The logistic fit function of sigmoidal shape was obtained by MÜet al. for relation between the peak areas of fluorescent zones and the log of estradiol amount in the HPTLC–Yes assay. 48 This new bioautographic method was introduced for detection of estrogenic compounds directly on a HPTLC plate by yeast cells with an estrogen receptor. Estrogenic substances induced production of an enzyme, which could be detected after incubation of the plate by spraying a proper substrate onto the plate. The fluorescence intensity of "estrogenic" zones was measured using a TLC scanner.
Overuse of antibiotics in human and veterinary medicine as well as in animal husbandry are considered as the main reasons for the rise of drug-resistant bacteria. Antibiotic residues or their metabolites in food should be below the maximum residue levels (MRLs) established by the European Union or below the safe levels imposed by the US Food and Drug Administration. TLC-bioautography is one of the screening techniques used in monitoring of antibiotic residues in food and feed.49,50 In our laboratory matrix solid-phase dispersion (MSPD) based on siliceous sorbents was combined with TLC-direct bioautography and HPLC for screening of enrofloxacin and ciprofloxacin residues in milk.51,52 Figure 2 presents the bioautogram of four samples (one blank and three containing antibiotics) and standards. The samples were the eluates from MSPD cartridges (2g Chromosorb WAW + 1 mL of spiked milk + 1 mL ACN) made in triplicates. In the instance of a blank sample the procedure was the same but performed for net milk. Figure 3 presents the similar bioautogram but for milk spiked at the MRL level (0.05 ppm of each antibiotic). The same samples were injected in parallel into the HPLC system. Quite good correlation between the results was found. The great advantage of TLC–DB compared to HPLC was a possibility of testing many samples at the same time.


Figure 3: TLC-DB of the eluates referring to the milk spiked at 0.05 ppm level. The level of ciprofloxacin and enrofloxacin in the MSPD eluates was 0.5 ppm each. Upper spots, enrofloxacin, lower spots, ciprofloxacin. Samples applied with Linomat 5 Camag applicator (Muttenz, Switzerland) in 50 õL volume. Three eluates from the MSPD cartridges at positions 4, 6 and 7. At other positions standards at 0.5 ppm level. Standards at 1 and 8 position were made in methanol, other samples were dissolved in HPLC mobile phase (water phase/ACN 70:30). The mobile phase for TLC was dichloromethane/ methanol/ 2-propanol/ 25% aqueous ammonia 3:3:5:2. The plate was developed to the end and then continuously for 2 hrs.
Irena Choma is a research worker in the Department of Chromatographic Methods of M. Curie-Sklodowska University in Lublin, Poland. She received her PhD in chemistry on chromatographic properties of modified porous glasses in 1990. Currently, her research focuses on the analysis of antibiotic residues in milk.
References
1. B.F. Brehm-Stecher and E.C. Johnson, Antimicrob. Agents Chemother., 47(10), 3357–3360, (2003).
2. Association of Official Analytical Chemists. Changes in Methods., J.Assoc.Off.Anal.Chem., 65, 466–467, 1982.
3. L.M. Kelly, M.R. Jacobs and P.C. Appelbaum, J. Clin. Microbiol., 37(10), 3296–3299 (1999).
4. FDA/CFSAN, Bacteriological Analytical Manual On-line, Chapter 20A Inhibitory substances in Milk, 2001. www.cfsan.fda.gov/~ebam
5. B. Tepe et al., Food Chemistry, 84, 519–525 (2004).
6. Association of Official Analytical Chemists. Official Methods of Analysis, 14th ed., secs 16.163; 42.299–42.303. AOAC, Arlington, Virginia, USA.
7. A. Shitandi and G. Kihumbu, Afr. J. Biotechnol., 3(1), 82–87 (2004).
8. T.A. Davies et al., J. Clin. Microbiol., 38(4), 1444–1448 (2000).
9. R.R. Goodall and A.A. Levi, Nature, 158, 675–676 (1946).
10. R. Fisher and H. Lautner, Arch. Pharm., (Weinheim), 294, 1 (1961).
11. B.J.R. Nicolaus, C. Coronelli and A. Binaghi, Farmaco (Pavia), Ed. Prat., 16, 349 (1961).
12. V. Betina, J. Chromatogr.,78, 41–51 (1973).
13. E. Meyers and D. Smith, J. Chromatogr., 14, 129–132 (1964).
14. Narasimhachari and S. Ramachandran, J. Chromatogr., 27, 494 (1967).
15. G.H. Wagman and J.V. Bailey, J. Chromatogr.,41, 263–264 (1969).
16. L. Williams and O. Bergersen, J. Planar Chromatogr.,14, 318–321 (2001).
17. D. Tasdemiret al., Pharmaceutical Biology, 42 (4–5), 374–383 (2004).
18. L. Zheng et al., World J. Microbiol.&Biotechnol., 21, 201–206 (2005).
19. G. Schmourlo et al., J. Ethnopharmacol., 96, 563–568 (2005).
20. K. Hostettman and A. Marston, Pure & Appl. Chem., 66 (10–11), 2231–2234 (1994).
21. A.I. Homans and A. Fuchs, J. Chromatogr., 51, 327–329 (1970).
22. B.M. Lund and G.D. Lyon, J. Chromatogr., 110, 193–196 (1975).
23. W.G. van der Sluis et al., J. Chromatogr., 214, 349–359 (1981).
24. G. Horváth et al., J. Planar Chromatogr.,17, 300–304, (2004).
25. C. Weins and H. Jork, J. Chromatogr.A , 750, 403–407 (1996).
26. G. Eberz et al., Chromatographia,43 (1–2), 5–9 (1996).
27. N. Islam et al., Pharmaceut. Biology, 41 (8), 637–640 (2003).
28. L. Rahalison et al., Phytochem. Anal., 2, 199–203 (1991).
29. M.M. Mackeen et al., 57c, 291–295 (2002).
30. E. Tyihák et al., in: Proc. Intern. Symp. on Planar Separations, Planar Chromatography 2001, New Milestones in TLC, ed. Sz. Nyiredy, Res. Inst. Med. Plants, Budakalász, 2001, p. 3–13.
31. E. Tyihák et al., Chem. Anal. (Warsaw),48, 543–553 (2003).
32. á. Moricz et al., J. Planar Chromatogr., 16, 417–420 (2003).
33. E. Tyihák et al., J. Planar Chromatogr., 17, 84–88 (2004).
34. R. Eymann and H.E. Hauck, in Proc. Intern .Symp. on Planar Separations, Planar Chromatography 2000, ed. Sz. Nyiredy, Res. Inst. Med. Plants, Budakalász, 2000, p. 67–75.
35. Chrom Biodip® Antibiotics1.05638 (Merck, Darmstadt, Germany). Text Chrom Biodip® leaflet, 33170206.doc, 2000
36. L. Botz, S. Nagy and B. Kocsis, Detection of microbiologically active compounds, in Planar Chromatography, ed. Sz. Nyiredy, Springer, Budapest, Hungary, 2001, p. 489–516
37. E.F. Queiroz et al., J. Chromatogr. A, 974, 123–134 (2002).
38. K. Hostettman et al., J. Planar Chromatogr.,10, 251–257 (1997).
39. J.L. Rios, M.C. Recio and A. Villar, J. Ethnopharmacol.,23, 127–149, (1988).
40. S. Nagy, et al., in: Proc. Intern. .Symp. on Planar Separations, Planar Chromatography 2001, New Milestones in TLC, ed. Sz. Nyiredy, Res. Inst. Med. Plants, Budakalász, 2001, p. 173–179.
41. S. Nagy et al., J. Planar Chromatogr., 15, 132–137 (2002).
42. S. Nagy et al., J. Planar Chromatogr., 16, 121–126 (2003).
43. I. Choma, A. Choma and K. Staszczuk, J. Planar Chromatogaphy, 15, 187–191 (2002).
44. I. Choma, A. Choma and K. Staszczuk, J. Liq. Chromatogr. & Rel. Technol., 25 (10–11), 1579–1587 (2002).
45. I. Choma et al., J. Liq. Chromatogr .& Rel. Technol.,27 (13), 2071–2085, (2004).
46. J. Kádár Pauncz and I. Harsányi, J. Chromatogr.,195, 251–256 (1980).
47. A. Ramirez et al., J. Chromatogr. B, 784, 315–322 (2003).
48. M.B. Müller et al., Chromatographia, 60, 207–211 (2004).
49. J.L Gafner, J. Assoc. Off. Anal. Chem. Int.82, 1–8 (1999).
50. P.K. Markakis, J. Assoc. Off. Anal. Chem.,79, 375–379 (1996).
51. I. Choma and I. Komaniecka, J. Liq. Chromatogr .& Rel. Technol, accepted for publication.
52. I. Choma Proc. Intern .Symp. on Planar Separations, Planar Chromatography 2005, ed. Sz. Nyiredy, Res. Inst. Med. Plants, Budakalász, 2005. p.279.














Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).

