Standardizing Proteomics: Results of a Collaborative Study
LCGC North America
A multilaboratory collaborative study organized by the Human Proteome Organization demonstrated that participating laboratories had difficulty in identifying components of a simple protein mixture.
The previous installment of this column (1) surveyed the challenges in obtaining high quality results in bottom-up proteomics, the sources of variability in proteomics experiments, and the difficulty in comparing results obtained from different laboratories using different sample preparation procedures, different instrument platforms, and different bioinformatic software. Five organizations were identified that have programs in place for standardizing proteomics workflows. These are the Association of Biomolecular Research Facilities (ABRF), the Biological Reference Material Initiative (BRMI), Clinical Proteomic Technology Assessment for Cancer (CPTAC), the Fixing Proteomics Campaign, and the Human Proteome Organization (HUPO). At the time of writing, the HUPO Test Sample Working Group had completed a collaborative study on protein identification but the results were not published until after the column had gone to press (2). This installment of "Directions in Discovery" will review the results of the study, as they clearly reveal the sources of variability in bottom-up proteomics and point to the road ahead in standardizing proteomics workflows.



Tim Wehr
The HUPO Test Sample
The HUPO sample consisted of 20 human proteins in the mass range of 32–110 kDa. To create the sample, candidate sequences were selected from the open reading frame collection and the mammalian gene collection, expressed in E. coli, and purified using preparative sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) or 2D high performance liquid chromatography (HPLC) (anion-exchange and reversed-phase chromatography). Purity of the proteins was determined to be 95% or greater by 1D SDS-PAGE. Quality and stability of the test sample was confirmed by mass spectrometry (MS) analysis. All of the 20 proteins were selected to contain at least one unique tryptic peptide of 1250 ±5 Da, each with a different amino acid sequence. This feature was designed to test for peptide undersampling derived from the data-dependent acquisition methods used by most bottom-up LC–MS protocols.
Sample Distribution to Collaborators
The 20-component test sample was distributed to 27 laboratories selected for their expertise in proteomics techniques. Of these, 24 were academic or industrial research laboratories or core facilities, while three were instrument vendors. Sample recipients were instructed to identify all 20 proteins and all 22 unique peptides with mass 1250 ±5 Da and to report results to the lead investigator of the Test Sample Working Group. Participants were allowed to use procedures and instrumentation they routinely employed in their laboratories so that effectiveness of different workflows could be assessed. To minimize variability in data matching and reporting, participants were requested to use the same version of the NCBI nonredundant human protein database.
Initial Study Results
In the initial reports returned to the Test Sample Working Group, only seven of the 27 participating laboratories identified all 20 proteins. The remaining 20 laboratories experienced a variety of problems. The first group (seven laboratories) reported naming errors in the protein identifications. The second group (six laboratories) reported naming errors, false positives, and redundant identifications. The remaining group of seven laboratories experienced several problems. These included trypsinization problems, undersampling, incomplete matching of MS spectra due to acrylamide alkylation, database search errors, and use of overly stringent search criteria.
Results for the peptide sequences were even more problematical; only one of the 27 laboratories reported detection of all 22 peptides. Six of the 22 peptides contained cysteine residues, which are modified in the reduction and alkylation steps performed before trypsin digestion. Only three additional laboratories reported detection of any of the cysteine-containing peptides. Several laboratories incorrectly reported 1250-Da peptides arising from contaminating proteins or missed trypsin cleavage.
Transfer of Data to Tranche and PRIDE
To facililate centralized analysis of study data, participants were asked to submit their results to Tranche. Tranche, in use since 2006, is a free, open-source file-sharing tool that enables collections of computers to easily share data sets and can handle very large data sets. Tranche is structured as a peer-to-server-to-peer distributed network. For the HUPO study, submitted information included raw MS data, methodologies, peak lists, peptide statistics, and protein identifications. After submission to Tranche, a copy of all data was transferred to PRIDE. PRIDE (PRoteomics IDEntifications) is a centralized, standards- compliant public data repository for proteomics data. It was designed to provide the proteomics community with a public repository for protein and peptide identifications together with supporting evidence for the identifications.

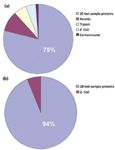
Figure 1: Number of tandem mass spectra assigned to tryptic peptides. Comparison of protein abundance from the centralized analysis of raw data collected from the participating laboratories (a) before and (b) following removal of individual laboratory contaminants. Adapted from reference 2.
Centralized Analysis of Study Data
Following downloading to Tranche, the centralized data was analyzed collectively to assign probabilities to identifications, determine total number of assigned tandem MS spectra, number of distinct peptides, and amino acid coverage. Inspection of the raw data revealed that the majority of participating laboratories had generated data of satisfactory quality to identify all 20 proteins and most of the 22 1250-Da peptides. Centralized data analysis provided several additional insights:
- The 20 human proteins accounted for 79% of the assigned tandem mass spectra, while contaminants account for 21% (Figure 1a). Contaminants included E. coli proteins, trypsin, and keratin.
- All 22 of the 1250-Da peptides were observed in only four laboratories.
- Laboratories using Fourier-transform ion cyclotron resonance (FTICR) mass spectrometers reported the highest number of assigned tandem mass spectra.
- E. coli proteins were found by all but two laboratories and most likely represented contaminants present in the sample.
- Tandem mass spectra assigned to keratins were primarily associated with laboratories using 1D PAGE; laboratories using chromatographic techniques had fewer keratin mass spectra.
- A subset of 15 laboratories reported proteins that probably represent contamination from high-abundance proteins commonly used in standardization (for example, bovine serum albumin and casein).
- When laboratory-introduced contaminants were excluded, 94% of the tandem mass spectra were accounted for by the 20 analyte proteins and the remaining 6% were assigned to E. coli proteins (Figure 1b).
- Tandem mass spectra matching the 1250-Da peptides were variable for each of the 20 proteins and were variably detected in the centralized analysis (Figure 2).

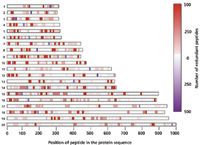
Figure 2: Peptide heat map representation for each of the 20 proteins from the centralized analysis of raw data from participating laboratories, showing frequency of observation of a given peptide and its position in the protein sequence. Red tones: redundant tryptic peptides excluding 1250-Da peptides; purple tones: redundant 1250-Da peptides. Adapted from reference 2.
Implications for the Proteomics Community
This study demonstrated that, even with a simple mixture of 20 proteins, the majority of the participating laboratories had difficulty in correctly identifying the components. Centralized analysis of the data revealed that these laboratories had generated tandem MS data of sufficient quality to identify all of the proteins and most of the 1250-Da peptides. It also identified database problems as a major source of error. Due to the construction of the database, the search engines employed by participants were unable to differentiate between multiple identifiers for the same protein, and manual curation of MS data was needed for correct reporting. The Working Group noted that search engines employed different algorithms for calculation of molecular weight and recommended that a common method be adopted. The study organizers provided additional recommendations based upon the results of the study:
- A simple sample such as the 20-protein mixture can serve as standard for benchmarking laboratory performance. Comparison of individual laboratory results with a centralized data repository can be used to identify specific problems with an individual workflow or instrument platform.
- Laboratories need to identify and monitor environmental contaminants such as keratin or carry-over species from prior experiments.
- Reporting of false discovery rates using decoy databases should be mandatory.
- Unique peptides and tandem MS spectra need to be monitored to address the issue of redundant identifications (that is, sequence variants of the same protein).
- Tools need to be created to transform data (for example, raw data, peak lists, and peptide and protein lists) into standardized formats to enable submission to data repositories.
Conclusion
The HUPO Test Working Group study is distinct from other collaborative studies of protein identification (3). First, the component proteins each contained a peptide of similar size to test for the ability of the mass spectrometer to reproducibly sample precursor ions. Second, participants received feedback from the working group on technical problems encountered in the initial analysis, and recommendations for improvement. Third, the working group performed centralized analysis of the combined data sets, which permitted discrimination of factors related to data generation versus data analysis. There are three key outcomes of this study that are important for the proteomics community. First, it demonstrates that a variety of instruments and workflows can generate tandem MS data of sufficient quality for protein identification. Second, operator training and expertise are critical for successful proteomics experiments. Third, environmental contamination can compromise data quality, particularly for gel-based workflows. Good laboratory practice including analysis of controls and blanks is necessary. Fourth, variations in database construction and curation must be addressed to allow proteomics researchers to obtain consistent results.
The simple equimolar 20-protein mixture used in the HUPO study hardly represents the complexity of a typical proteomics sample, which can contain hundreds of thousands of analytes covering several orders of magnitude in abundance. However, it did serve to illuminate factors that compromise data quality and to provide guidelines for improving performance in proteomics studies.
Tim Wehr "Directions in Discovery" editor Tim Wehr is staff scientist at Bio-Rad Laboratories, Hercules, California. Direct correspondence about this column to "Directions in Discovery," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.
References
(1) T. Wehr, LCGC 27 (7), 558–562 (2009).
(2) A.W. Bell, E.W. Deutsch, C. E. Au, R.E. Kearney, R. Beavis, S. Sechi, T. Nilsson, J.J.M Bergeron, and the HUPO Test Sample Working Group, Nature Methods 6, 423–429 (2009).
(3) R. Aebersold, Nature Methods 6, 411–412 (2009).
















This information is supplementary to the article “Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing”, which was published in the May 2025 issue of Current Trends in Mass Spectrometry.

Investigating the Protective Effects of Frankincense Oil on Wound Healing with GC–MS
April 2nd 2025Frankincense essential oil is known for its anti-inflammatory, antioxidant, and therapeutic properties. A recent study investigated the protective effects of the oil in an excision wound model in rats, focusing on oxidative stress reduction, inflammatory cytokine modulation, and caspase-3 regulation; chemical composition of the oil was analyzed using gas chromatography–mass spectrometry (GC–MS).


