Split Peaks — A Case Study
LCGC North America
Case studies are good ways to look at specific examples of common liquid chromatography (LC) problems and to draw general conclusions that can be applied to prevent similar problems from happening for other workers. The example in this month's installment of "LC Troubleshooting" comes from a reader who works in the pharmaceutical industry. The sample is a cold-cough syrup analyzed with an ion-pairing LC method. I have disguised the details somewhat to protect the proprietary nature of the method, but there should be sufficient information to help us gain some knowledge of the peak-splitting problem experienced by the user.
Case studies are good ways to look at specific examples of common liquid chromatography (LC) problems and to draw general conclusions that can be applied to prevent similar problems from happening for other workers. The example in this month's installment of "LC Troubleshooting" comes from a reader who works in the pharmaceutical industry. The sample is a cold-cough syrup analyzed with an ion-pairing LC method. I have disguised the details somewhat to protect the proprietary nature of the method, but there should be sufficient information to help us gain some knowledge of the peak-splitting problem experienced by the user.


John W. Dolan
The Problem
The analyst was developing an assay for stability and purity of the product. After approximately 40 injections, she noticed that the codeine peak had deteriorated to a peak with a shoulder on it, as illustrated in Figure 1a. The peak splitting increased as the run continued, with a distinct doublet appearing after three more injections (Figure 1b) and showed further deterioration (Figure 1c) after another eight injections. When one is developing a stability-indicating assay, the appearance of new peaks can indicate that additional decomposition has occurred. The analyst suspected this and checked the spectra across the entire peak with a photodiode-array detector. All of the spectra were identical to that of codeine, which suggests that it is peak splitting, not chemical degradation of the sample or appearance of a new compound. Other peaks in the sample showed similar distortion.


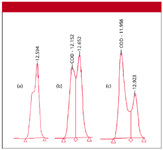
Figure 1: Codeine peak from a sample of cold-cough syrup (a) after approximately 40 injections, (b) three injections later, and (c) after eight more injections. See text for details.
The next step taken was to see if reference standards behaved the same as the sample. The peak for the reference standard shown in Figure 2a was obtained when the column was working well, whereas the peak in Figure 2b was obtained from a run made soon after that of Figure 1c. It is clear that the problem is not unique to the sample.


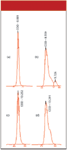
Figure 2: Codeine peak for injection of reference standard obtained (a) using a new column, (b) soon after the run of Figure 1(c), (c) using a replacement column, and (d) after failure of the replacement column. See text for details.
The Method
When I am presented with a problem such as this, I like to look carefully at the analytical method to see if any red flags appear. The method was ion pairing, using one of the sulfonic acid ion-pairing reagents at a concentration of 10 mM as the A solvent, with the pH adjusted to 2.8 with phosphate buffer. The B solvent was methanol. The isocratic portion of the run was 20% B. At the end of the run, the column was flushed with 60% methanol to remove late-eluted materials. The column was a 250 mm × 4.6 mm C18 "aqueous phase" column packed with 5-mm particles and thermostated to 40 °C with a flow rate of 1 mL/min.
The sample was a suspension that contained approximately 10 components. In addition to the codeine and other active ingredients, the sample contained 30% of an artificial sweetener plus a thickener. The sample was diluted 10-fold with 90:10 A:B solvents and 20 μL was injected. This resulted in an injection of approximately 2 μg of codeine and 600 μg of the sweetener; the concentration of thickener was not indicated.
So what looks like a problem? To me, the method looks all right. The ion-pairing reagent concentration and pH are reasonable, as are the other operating conditions. The column is produced by a well-known manufacturer and uses type B silica that is more stable than older materials. With older, type A silica columns and column-packing techniques that were not as good as those used today, column stability with ion-pairing methods was poorer than standard reversed-phase methods. However, I have not heard of stability problems with type B columns and ion pairing. I'm not sure how the C18 "aqueous phase" column is affected by ion pairing. This column technology is proprietary, but often uses low bonded-phase coverage or polar endcapping to avoid phase dewetting with 100% water. There is nothing about this chemistry that should be inherently unstable under ion-pairing conditions.
The sample raises more questions to me. A suspension means that all of the sample components are not fully soluble. This can result in insoluble materials being injected onto the column. I suspect that this is the root cause of the problem. Overload is another possibility. As a general rule of thumb, one can assume the column will not be overloaded until more than approximately 10 μg of sample is injected per gram of packing material. A 250 mm × 4.6 mm column contains approximately 2.5 g of packing, so it should tolerate about 25 μg without overload problems. An injection of 2 μg of codeine should not be a problem, but the artificial sweetener certainly overloads the column at 600 μg on-column. The thickener is likely to increase the viscosity of the sample, which can result in some peak broadening. However, the reference standard behaved the same as the sample, so it appears that the presence of the sweetener and thickener were not causing the problem with the injections of sample.
Attempts to Fix the Problem
The first attempt to correct the problem was to clean the column according to the manufacturer's instructions. Generally, this is a wash with 100% methanol or acetonitrile to remove strongly retained materials. This did not correct the problem.
Next, the column was reversed and back-flushed. This corrected the problem for a while, but the problem reappeared. By this time, the column pressure had increased by about 300 psi. After the problem appeared again, the analyst removed the fitting at the column inlet. Neither the frit nor the packing material appeared to be discolored, and no void was evident.
Next, the column was replaced with a new one, and the problem disappeared. The codeine peak on the new column (Figure 2c) looked just as good as it did on the previous column (Figure 2a). Ultimately, however, the second column deteriorated (Figure 2d), although the back-pressure did not increase.
Likely Cause
To me, all of the symptoms point to a blocked frit at the column inlet. The classic symptom of a blocked frit or column void is doubled peaks for all peaks in the chromatogram. I think of this as something that distorts the sample as it is introduced to the column so that its initial shape is disturbed. Once this distortion takes place, it will not be corrected under isocratic conditions. Because distortion happens before any separation, it affects all peaks in a similar manner. Sometimes, one can see particulate matter when inspecting the frit, but the internal pores in the frit can become blocked without any visible change in the surface, so a visual inspection might not be very meaningful. Further evidence of a blocked frit is the rise in column pressure and the correction of the peak distortion problem when the column was reversed. Reverse flushing often displaces particulate matter from the frit, but it doesn't always work.
If the problem indeed is due to collection of particulate matter on the frit, where is it coming from? The logical source is the insoluble materials in the sample suspension. Furthermore, if the sample components are not soluble in an aqueous formulation, it is quite possible that further solubility problems will occur when the sample contacts the 20% methanolic mobile phase. This can be tested visually by adding a few drops of mobile phase to a test tube containing the diluted sample. Any precipitation or cloudiness indicates that solubility will be a problem.
Corrective Action
The obvious way to prevent particulate matter from blocking the column inlet frit is to remove it from the sample before injection. There are four ways to do this. First, and perhaps the most obvious, is to filter each sample before injection. Disposable syringe filters are available with 0.5- or 0.2-μm porosity. The 0.5-μm filters should be sufficient to remove any particulate matter that would cause column problems. The 0.2-μm filters generally are reserved for removal of bacterial contamination and are difficult to use because they are prone to blockage. When sample filtration is to be performed, one needs to make sure that the filter does not selectively remove something from the sample (besides the particles) and that it does not add unwanted contaminants to the sample. A third problem with filters is that there is always some holdup volume in the filter so some sample volume is lost, which can be a problem with low-volume samples. So if one desires to use sample filtration to ameliorate the problem, some kind of validation process must be undertaken. All of this extra work, plus the added cost of the filters, makes sample filtration an undesirable choice in many laboratories. An alternative to sample filtration is one that is used on every sample in my laboratory — just spin the samples in a centrifuge to remove particulate matter and transfer the supernatant to the sample vial. This can be done in individual vials or in 96-well plate formats.
A third technique to remove particulate matter is to use a guard column. Guard columns are just miniature versions of the analytical column that are designed to trap the chemical and physical debris that can cause problems at the head of the analytical column. Many workers find guard columns both cost effective and a simple way to solve problems such as the present one. The guard column is replaced daily or after a predetermined number of runs so that the collected material does not bleed through onto the analytical column.
Another tool that usually is sufficient to prevent problems such as the present one is to use an in-line filter containing a 0.5-μm porosity frit. This will trap anything that would otherwise get trapped at the head of the column. When a pressure rise is observed, just replace the frit and you should be back in business. In my laboratory, we mount an in-line filter on every instrument just downstream from the autosampler and centrifuge all samples before injection. This greatly reduces column failure from high back-pressure.
What about column back-flushing? For most columns packed with 5-μm particles, the column can be reversed and operated in either direction. The frits at the inlet and outlet of 5-μm columns usually are 2-μm porosity and hold the packing particles in place in either flow direction. For 3-μm particle size columns, however, the 2-μm frit is too porous to use at the column outlet, so 0.5-μm porosity outlet frits are used on 3-μm columns. These frits are much more prone to blockage and result in an increase rate of inlet-frit blockage when 0.5-μm frits are used on the column inlet. Some manufacturers alleviate the problem by using a 2-μm frit on the column inlet and an 0.5-μm frit on the outlet. Columns with this frit arrangement cannot be reversed. Whereas my advice for years has been that there are no problems with reversing columns, I now strongly suggest that you read the column care and use instructions to see if column reversal is permissible for your particular column.
Conclusions
So what have we learned from this case study? Don't inject samples that are likely to contain particulate matter! If you suspect a blocked frit, column reversal often will correct the problem. However, if you don't correct the root cause of the problem, it will occur again, as was the case in the example presented here. The simplest way to avoid injecting particulate matter is to centrifuge all samples to remove as much of the insoluble material as possible and then use a 0.5-μm porosity in-line filter to catch the occasional bit of particulate matter that is missed or originates from worn pump seals or injector rotors.
The appearance of split or distorted peaks for all peaks in the chromatogram is evidence that the sample was not introduced onto the column in a symmetric manner. The most common cause is a blocked frit or a void at the head of the column.
John W. Dolan "LC Troubleshooting" Editor is Vice-President of BASi Northwest Laboratory of McMinnville, Oregon; a Principal Instructor for LC Resources, Walnut Creek, California; and a member of LCGC's editorial advisory board. Direct correspondence about this column to "LC Troubleshooting," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail John.Dolan@Bioanalytical.com.
For an ongoing discussion of LC trouble-shooting with John Dolan and other chromatographers, visit the Chromatography Forum discussion group at www.chromforum.com.
















SPE-Based Method for Detecting Harmful Textile Residues
January 14th 2025University of Valencia scientists recently developed a method using solid-phase extraction (SPE) followed by high-performance liquid chromatography coupled to high-resolution mass spectrometry (HPLC–HRMS/MS) for detecting microplastics and other harmful substances in textiles.





