The Role of the Signal-to-Noise Ratio in Precision and Accuracy
Improvement of detection and quantification limits for LC methods is a result of taking advantage of all the tools you have at your disposal.
The minimum peak size for reportable results for a liquid chromatographic (LC) method depends upon the application and the signal-to-noise ratio (S/N). The signal for the analyte peak and the baseline noise both contribute to S/N. This month's instalment of "LC Troubleshooting" takes a look at the measurement of S/N, some techniques for determining the detection limits for a method, and some tricks to improve S/N.
Determining S/N
With today's data systems, S/N is often a number calculated as part of a data report for an analyte and can be based upon a mathematical treatment of the data, such as a root-mean-square determination of noise. Manual measurement of S/N can be performed, as illustrated in Figure 1. Select a section of baseline free of peaks, expand the plot scale so that the noise is easy to determine, and draw two lines tangent to the noise. The vertical distance between the two lines is the noise for the chromatogram. The signal is measured from the midpoint of the noise to the top of the peak. S/N is obtained by dividing the signal by the noise. Use whatever measurement units are convenient — millivolts, absorbance units, millimetres — they will cancel out and leave a unitless quantity.


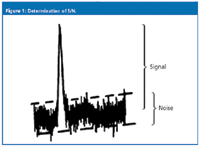
Figure 1: Determination of S/N.
S/N and Method Performance
Most readers of this column work for the pharmaceutical industry, in which sample types fall into two major categories. The first is pharmaceutical analysis, which focuses on analysis of the active pharmaceutical ingredient (API) in a standard (called the drug substance) or in a formulated product (called drug product analysis). In instances for which the analysis is for quantification of the API in either drug substance or product, precision and accuracy limits in the 1–2% range are required. (It should be noted that strictly speaking, we are talking about imprecision and inaccuracy, but common usage prevails for the terms precision and accuracy for these parameters.) The limits are more lenient for impurities, degradants and minor components in the same samples. The second type of analysis is measurement of the drug in a biological sample, such as plasma, and is called bioanalysis (or just bioanalytical). Bioanalytical work has a much wider tolerance than pharmaceutical analysis — 15–20% precision and accuracy in most cases. (Similar precision and accuracy can be acceptable for trace analysis of pharmaceutical impurities or degradants.) This increased allowance for method variation is because S/N in bioanalysis is much smaller than pharmaceutical analysis as a result of lower drug concentrations and often more background.
If the acceptable method precision differs by an order of magnitude between bioanalytical and pharmaceutical analysis, it is obvious that an acceptable peak in one application can be unacceptable in another. It really has to do with the method's tolerance for error and S/N. I like to use a rule of thumb to help me determine how small a peak can be and still generate usable data:


where %RSD is the percent relative standard deviation (sometimes called the coefficient of variation [CV]). Equation 1 gives us an idea of what S/N is required for a desired method precision. For example, if the %RSD for a method's precision can be no larger than 2%, this means that S/N = 25 is required (this assumes that S/N is the primary contribution to %RSD, which might or might not be true). For bioanalytical methods, S/N = 2.5 would translate into %RSD ≈ 20%.
Let's see how S/N relates to limits of detection and quantification. Some workers consider S/N = 3 to be the limit of detection (LOD) for a method and others use S/N = 10. From Equation 1, this converts roughly to 15 and 5% RSD, respectively. I know of companies that define the LOD as the sample that generates peaks ≤ 30% RSD and limit of quantification as ≤ 10% RSD. These would correspond to S/N ≈ 1.7 and 5, respectively. It is clear that the criterion that determines the lower limits of a method can vary by an order of magnitude, depending upon the user and the application. The advantage of using S/N to define the LOD and lower limit of quantification (LLOQ) is that such determinations can be based upon a single injection, rather than several injections required for a statistical result. Thus, one can confirm S/N quickly during a system suitability test without running many samples.
For the 1–2% precision and accuracy required by pharmaceutical methods for potency, it can be seen that large S/Ns are needed. Fortunately, for this type of method, the chemist is rarely sample limited, so injection of a higher concentration sample is usually convenient. For methods in which impurities or degradants must be reported, one generally needs to quantify any peaks larger than 0.1% of the main peak. What kind of confidence can you place in the results for peaks at this level? Some rough calculations allow us to predict precision for a method using UV detection.
UV detectors are specified to be linear to 1.0 absorbance unit (AU), and some models are linear for larger signals. If we inject a peak that is 1.0 AU tall, what S/N can be expected at the 0.1% level? A typical UV detector specification is for noise of ±5 X 10-5 AU with a dry flowcell. With mobile phase flowing through the cell, one should be able to get within about 10-fold of this value, let's say 5 X 10-4 AU. But 0.1% of 1 AU is 1 X 10-3 AU — about twice the noise level, not S/N > 10, so Equation 1 suggests RSD > 25% for these conditions. And a noise level this low might not be reasonable with a real sample. Is it a hopeless situation? Perhaps it is if we use peak height for quantification, but use of peak area will generally solve the problem. Although peak height and peak area should be proportional, by the time a peak is large enough to be 1 AU tall, it is often exhibiting some column overload. In such situations, the peak is broadened from overload, so the area increases faster than the height. This means that an impurity at 0.1% of the peak area of a 1.0 AU tall peak will be taller than 1 X 10-3 AU. Even so, it requires quiet baselines to quantify peaks at the 0.1% level.
Improving S/N
The previous discussion makes it clear that low detection and quantification limits require S/Ns consistent with the method precision and accuracy requirements. The only way to lower the LOD and LLOQ is to reduce the noise, increase the signal or both. Let's look at some of the options available.
Reduce noise: Reduction of baseline noise can be accomplished in several ways. These include the following.
- Signal averaging: The detector time constant and data system sampling rate determine the amount of smoothing that takes place for the signal. General guidelines are to set the detector time constant to about one-tenth of the width of the narrowest peak of interest. Similarly, the data system generally defaults to 10–20 data points across the peak. Larger time constants and slower data rates average the signal more, resulting in a reduction of noise, but excessive averaging will reduce the signal for peaks of interest as well. However, changing these values from the defaults can reduce noise without compromising the signal — it is worth checking.
- Temperature control: Variation of the column temperature and the temperature of the mobile phase entering the detector can create undesirable noise. For best results, use a column heater, insulate the tubing connecting the column and detector, and protect the detector from drafts. Although the overall laboratory temperature can be constant, the local temperature can vary significantly, especially if the LC system is near a heating vent.
- Reagent and solvent purity: Use high performance liquid chromatography grade (HPLC-grade) solvents for the lowest background signals. Also use high-purity reagents for sample preparation. Match the injection solvent with the mobile phase.
- Additional pulse damping and mixing: Most LC systems are plumbed to obtain small system dwell volumes (the volume between the solvent mixer and the head of the column). Additional mixing volumes and pulse-dampening devices will often reduce baseline noise, but at the expense of increased dwell volumes. Dwell volume differences can be a problem for transfer of gradient elution methods, but only affect system washout and solvent changeover for isocratic methods. Improved mixing for gradient methods can often be obtained by premixing the A and B solvents. For example, a 5–95% gradient will generally have a quieter baseline than the pure solvents are mixed on-line if 5% B is mixed into A and 5% A into B with a gradient setting of 0–100%. Isocratic methods will have quieter baselines if the solvents are mixed manually.
- Sample clean-up and column flushing: Sample clean-up steps will reduce the amount of extraneous material that gets introduced onto the column and will generally result in lower baseline noise. If the column is flushed with strong solvent at the end of each run, strongly retained materials will be eluted from the column, and it will reduce background noise.
Increase signal: Increase of the analyte signal while maintaining a constant noise value will increase S/N. When considering trace analysis and detection limits, peak height is a more important parameter than peak area, so anything that can be done to increase the peak height is generally beneficial.
- Wavelength selection: For UV detection, operating at the maximum absorbance for each peak will maximize the signal. Many methods are set-up with a wavelength chosen as a compromise for the response of all sample components, so one or more components can give stronger signals if another wavelength is chosen. Nearly all modern detectors are under the control of software, so it is easy to change the detection wavelength during a run so that the optimum wavelength is used for each peak. All organic compounds have a strongly increasing absorbance as the wavelength is reduced below 220 nm, so lower wavelengths often give larger signals than higher wavelengths. However, background noise and response of interfering peaks is likely to increase at low wavelength too, so it is a good idea to evaluate the overall gain in S/N when a change in wavelength is made.
- Better detector or modify analyte: The UV detector is the most popular detector for LC work, but several other detectors are available that offer increased selectivity. For example, a fluorescence detector can produce a large signal increase and a reduction in the background noise for samples that fluoresce or that can be derivatized to fluoresce. Electrochemical (amperometric) detectors and mass spectroscopic detectors are other examples of detectors that can give huge increases in signal for some compounds without the same increase in noise.
- Inject more sample: In many methods, sample availability is not a limiting factor, so a larger mass of sample can be injected to increase the analyte signal. One can take advantage of on-column concentration for both isocratic and gradient methods by using an injection solvent that is weaker than the mobile phase to enable injection of much larger sample volumes than are used normally. If you increase the sample mass on column, remember to check to be sure that large peaks are not overloaded.
- Reduce the peak width: For a given peak area, narrower peaks will be taller, thus improving detection limits. Volumetric peak widths can be reduced in isocratic methods by reducing the retention time (use of stronger mobile phase). With gradient methods, steeper gradients will reduce peak widths. Other tricks are to use smaller particle columns, shorter columns and narrower internal diameter columns, either alone or in combination. If you go down this route, the detector time constant and data system collection rate might need to be modified (see the previous discussion on reduction of noise).
Conclusions
Improvement of detection and quantification limits for LC methods is a result of taking advantage of all the tools you have at your disposal. Increasing the analyte signal size, reducing the baseline noise or a combination of both will be required to get lower method limits.
"LC Troubleshooting" editor John W. Dolan is vice-president of BASi Northwest Laboratory of McMinnville, Oregon, USA; a training consultant for Rheodyne LLC, the LC Resources Training Group, of Walnut Creek, California, USA; and a member of the Editorial Advisory Board of LC•GC Europe. Direct correspondence about this column to "LC Troubleshooting," LC•GC Europe, Advanstar House, Park West, Sealand Road, Chester, CH1 4RN, UK.
Readers can also direct questions to the on-line Chromatography Forum at http://www.chromforum.com







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")




. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

