Reversed-Phase Liquid Chromatography and Water, Part 1—How Much is Too Much?
LCGC Asia Pacific
When can we use completely aqueous eluents with reversed-phase stationary phases, and what happens if we make a mistake?
When can we use completely aqueous eluents with reversed-phase stationary phases, and what happens if we make a mistake?
Reversed-phase liquid chromatography (LC) is an incredibly powerful mode of separation that is applicable to a wide variety of applications ranging from the separation of small organic acids to 150 kDa proteins. Reversed-phase separations have limitations, however, with one of the most practically significant ones being low retention for compounds that are highly water soluble (that is, hydrophilic). Understanding the general trend for reversed-phase separations that retention increases as the fraction of water in the eluent increases, and encountering situations where retention is too low for an analyte of interest pushes us to use eluents with higher and higher levels of water. This, then, leads to the question,“How much water is too much?” Jumping to the end of this article, the short answer will be “It depends”.
In some cases, using completely aqueous eluents (that is, containing no organic solvent such as methanol or acetonitrile) may be completely acceptable and in fact can provide very useful separations of highly hydrophilic molecules. In other cases, completely aqueous eluents can cause some reversed-phase stationary phases to behave in undesirable ways and should be avoided. This is not a new topic by any means (1,2), but the options we have for handling such situations are constantly changing. It is worth taking the time here to refresh our perspective on the topic as column manufacturers introduce new stationary phase chemistries and particle morphologies, and our knowledge of what goes on inside the column improves through fundamental research. In this first segment on this topic, I will review the basic concepts that are important for reversed-phase separations in highly aqueous eluents, summarize recent advances in our understanding of what goes on inside the column, and provide examples of bad column behaviour and potential remedies.
Basic Concepts for ReversedâPhase Separations with Aqueous Eluents
It has been observed since the early days of LC that operating a typical C18-type reversed-phase column in a completely aqueous eluent can lead to gradual or sudden decreases in retention time (3). For at least a couple of decades, the prevailing idea seems to have been that the observed decrease in retention time was due to a change in the conformation of C18 chains bonded to the particle substrate in aqueous eluents from highly extended chains (that is, perpendicular to the substrate surface), to ones that laid down on themselves (that is, parallel to the surface). The latter state was commonly referred to as “phase collapse” (1–3). These observations also led to the notion that operating reversed-phase columns in completely aqueous eluents was generally a really bad idea, so much so that this idea made it into the Top 10 Myths of LC addressed by Ron Majors just five years ago (4). Around the late 1990s, however, experimental evidence led a number of groups to adopt the idea that the decrease in retention time was due instead to “dewetting” of the stationary phase (5). Specifically, the idea is that the high surface tension of an aqueous eluent in contact with a hydrophobic surface causes the eluent to extrude from the pores of the stationary phase particle. If there is no liquid in the pores of the particle, this effectively reduces both the column dead volume (that is, the volume of mobile phase inside the column, Vm) and the volume of stationary phase that is accessible to the analyte. Indeed, significant decreases in Vm have been measured for C18âtype phases when switching from organicârich to completely aqueous eluents, and these decreases are correlated with decreases in retention observed for analytes of interest (3,6). The extent to which this extrusion of the eluent occurs under actual separation conditions depends on a number of factors, including the particle pore size, stationary phase chemistry, column temperature, and operating pressure (through this last factor, we could say dewetting depends on particle size and flow rate, as well).
For over a decade, Siepmann, Schure, and coworkers have been using Monte-Carlo molecular simulations to study the microscopic details of mobile and stationary phases in an effort to better understand how reversed-phase separations work (7). The results of these studies complement experimental work, and provide insights that cannot be obtained easily by experiment. Among a number of topics, they have addressed the question of what happens to reversed-phase stationary phases in aqueous eluents. They have found that the results of simulation support the idea that the observed decreases in retention must be due to loss of eluent from the particle pores, rather than physical collapse of the stationary phase chains onto themselves (8).
The equation of Young and Laplace is most commonly used to rationalize the effects of different chromatographic variables on the dewetting phenomenon (1,5,6,9). This equation, shown in equation 1 (10), provides a relationship between the pressure (P) required to force a liquid into a capillary, the contact angle of the liquid on the interior surface of the capillary (θ), the surface tension of the liquid (γ), and the radius of the capillary (r):



Since the contact angle of water on a hydrophobic surface, such as C18-modified silica, is greater than 90°, a positive pressure is required to force water into the pore of a particle. Under conditions typical of modern LC separations, the pressure required to push the eluent through the particle bed will exceed the pressure required to push the eluent into the pores of the particles at most points along the column length. However, when the flow is stopped (for example, if the LC is not operated overnight), an aqueous eluent can be spontaneously extruded from the pores of a hydrophobic stationary phase particle, and the retention behaviour will look very different the next time the column is used. With this framework in mind, we can think about how different chromatographic variables will affect this behaviour. As the stationary phase becomes less hydrophobic, the contact angle for water will decrease. When the angle is less than 90°, the pressure indicated by equation 1 becomes negative, meaning that the eluent will spontaneously be drawn into the pores of the particles. Among chromatographers, we refer to this as “wetting” of the pores. Equation 1 also suggests that the particle pore size ought to play a role (that is, through r), with the pressure required to force eluent into the pore increasing as the pore size decreases.
The Washburn equation provides a helpful framework for thinking about the effects of these parameters, but, of course, the pores of chromatographic particles are not ideal by any means. The pore structure itself is heterogenous, with a distribution of diameters and shapes, and the chemistry of the pore surface is locally heterogeneous with some unbonded silanol sites, stationary phase ligands (for example, C18), and endcapping functional groups (for example, trimethylsilyl groups). And so, we look to experimental results for the definitive answer to the question, “How much water is too much?” The chromatographic literature provides useful data that at least establish trends, even if the results are not exactly transferrable to a particular set of conditions of interest. For example, Bidlingmeyer and Broske showed results that speak to the effect of particle pore size, stationary phase chemistry, and column temperature on the extent to which dewetting occurs in aqueous eluents (11). They found that, for one type of C18 stationary phase, there was a retention loss of 80% for particles with a pore diameter of 80 Å, but, with a diameter of 150 Å and the same stationary phase, there was no measurable retention loss. On the other hand, they found that very little retention loss was measured for a phenyl-type stationary phase, even for particles with 80 Å pores. Walter and coworkers observed similar trends, and also described results for the dependence of retention loss on stationary phase bonding density, the concentration of methanol in the eluent, and use of a post-column restrictor to increase pressure inside the column (5).
Testing for Dewetting, and Some Remedies
One good way to assess whether or not a reversed-phase stationary phase is susceptible to dewetting under a particular set of conditions is by first equilibrating the column under conditions where the stationary phase is highly solvated, or fully wetted. For most reversed-phase stationary phases, this could be an eluent high in methanol or acetonitrile content. Flushing the column at a modest flow rate for a time equivalent to about 20 column volumes should be more than enough. Then, we switch to the aqueous eluent (or whatever eluent is useful for the application at hand), begin injecting a mixture of two or three probe compounds that have reasonable retention under these conditions, and monitor the change in retention factor (k) as the column equilibrates. One could simply wait for 20 column volumes to pass first before injecting the test sample, but, if we start injecting right away, we also learn something about how quickly the column equilibrates when switching from the organic-rich to the aqueous eluent. Then, once the retention has stabilized after this initial equilibration step, turn the flow off, wait 10 minutes, then turn the flow back on, and start injecting the test mixture again. If there is a significant difference between the retention before and after stopping the flow, dewetting is likely to be a serious problem under these conditions. Retention will vary from day to day, depending on the post-column flow restriction in the system, and peak shapes may deteriorate and become variable.
Figures 1 and 2 show representative results from such a test, conducted with two C18-type stationary phases that are otherwise very similar (that is, they use the same base silica), but one (AQ-C18) is designed for use in highly aqueous eluents. In other words, the AQ-C18 is engineered to avoid the dewetting phenomenon in completely aqueous eluents. Figure 1 shows the retention factor for uracil measured on these two columns in a completely aqueous eluent, injecting sample once per minute, before and after stopping the flow for 10 minutes. For the very first injection, the retention is slightly lower than the rest of the points, because the column has not equilibrated from the 50:50 organic–water flushing solvent to the completely aqueous eluent. After equilibration, the retention of uracil on the two phases is remarkably similar. After turning the flow off for 10 minutes, we observe that the two phases behave very differently. For the AQ-C18, there is no statistically significant change in the retention of uracil. On the other hand, the retention of uracil on the C18 column decreases by about 75%.

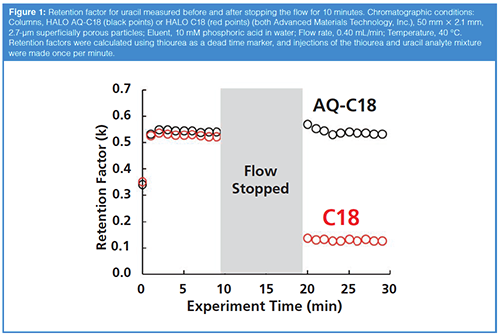
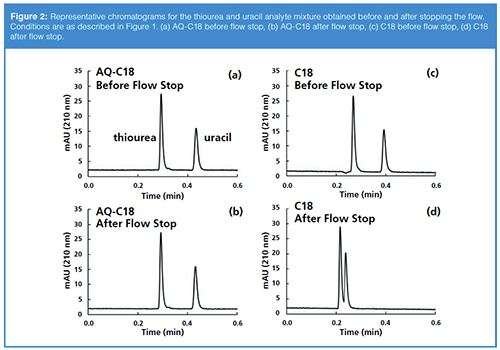
Representative chromatograms for the two columns before and after stopping the flow are shown in Figure 2. The chromatograms for the AQ-C18 column are indistinguishable, as expected. In the chromatograms for the C18 column, though, we see that there is not only a change in the retention factor of uracil, but also a 23% decrease in the measured dead time. This is consistent with the idea discussed above, that eluent is extruded from the pores of the stationary phase particles when dewetting happens, effectively decreasing the dead volume of the column.

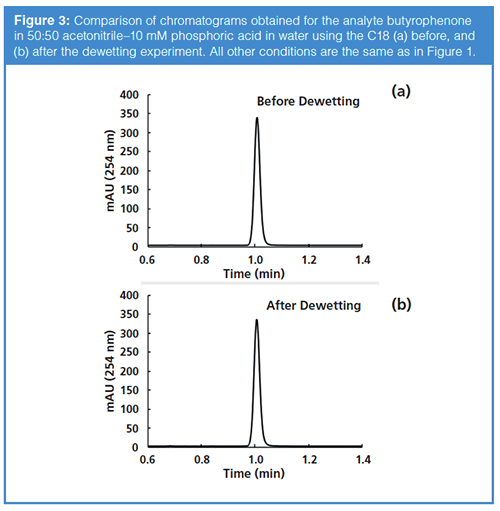
The good news is that dewetting does not have to be a death sentence for reversed-phase columns. As is the case when recovering a column that has dried out during storage (12), columns that have dewetted can be recovered by flushing with several column volumes of organic-rich eluent. Figure 3 shows a comparison of chromatograms obtained for the analyte butyrophenone before and after the dewetting experiment summarized in Figures 1 and 2. In this case, simply pumping 50:50 acetonitrile–water through the column at 0.4 mL/min for 5 minutes (pressure drop across the column was about 100 bar) was enough to restore both the retention and peak shape for this column to a “like new” state.

Summary
In this instalment of “LC Troubleshooting”, I have described the phenomenon that has become known as “dewetting” in reversedâphase chromatography. I have discussed the basic principles that explain why and when this occurs, so they can be used as a guide during method development. Since the extent of dewetting depends on a number of factors, including particle pore size, stationary phase chemistry, and operating conditions, it is a good idea to test for dewetting using the stop-flow during method development of applications involving an alkyl bonded phase (for example, C8 or C18), if they will be used with eluents containing less than 5% organic solvent. This piece has been restricted to isocratic conditions (that is, the use of completely aqueous eluents with reversed-phase stationary phases). In a future instalment, I will discuss the implications of using solvent gradient elution involving these highly aqueous eluents.
Acknowledgements
I’d like to thank Dr. Stephanie Schuster of Advanced Materials Technology for providing the columns used in this work, and Dr. Mark Schure and Dr. Richard Henry for helpful discussions on the topic of dewetting.
References
- M. Przybyciel and R.E. Majors, LCGC North Amer.20, 516–523 (2002).
- J.W. Dolan, LCGC Europe21(12), 624–627 (2008).
- P. McDonald, Advances in Chromatography (Marcel Dekker, New York, USA, 2003) pp. 323–375.
- R.E. Majors, LCGC Europe 26(10), 584–592 (2013); R.E. Majors, LCGC Europe 26(11), 630–636 (2013).
- T.H. Walter, P. Iraneta, and M. Capparella, J. Chromatogr. A1075, 177–183 (2005). DOI:10.1016/j.chroma.2005.04.039.
- K. Nakamura, H. Nakamura, S. Saito, and M. Shibukawa, Anal. Chem.87, 1180–1187 (2015). DOI:10.1021/ac503802q.
- M.R. Schure, J.L. Rafferty, J.I. Siepmann, and L. Zhang, LCGC Europe27(1), 18–27 (2014).
- L. Zhang, L. Sun, J.I. Siepmann, and M.R. Schure, J. Chromatogr. A1079, 127–135 (2005). DOI:10.1016/j.chroma.2005.03.124.
- M. Schure, N. Devitt, R. Moran, J.M. Godinho, and B. Wagner, J. Chromatogr A Submitted (2019).
- A.W. Adamson and A.P. Gast, Physical Chemistry of Surfaces (Wiley, New York, USA, 6th Ed., 1997).
- B.A. Bidlingmeyer and A.D. Broske, Journal of Chromatographic Science42, 100–106 (2004). DOI:10.1093/chromsci/42.2.100.
- D.R. Stoll, LCGC Europe30(7), 352–357 (2017).
Dwight R. Stoll is the editor of “LC Troubleshooting”. Stoll is a professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota, USA. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 60 peer-reviewed publications and three book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: LCGCedit@ubm.com







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


Distinguishing Alcohol- from Non-Alcohol-Associated Liver Cirrhosis with LC-MS
May 7th 2025A pilot study investigating whether nicotinamide adenine dinucleotide kinase (NADK) expression is selectively diminished in alcohol-associated liver cirrhosis (AC), as well as evaluating its potential as a biomarker for this condition, measured AC and non-AC (NAC). Nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+) levels in human liver samples were measured using liquid chromatography-mass spectrometry (LC-MS).






