Retention Modelling of Peptide and Protein Separations
The Application Notebook
Modelling of peptide and protein separations is investigated using commercially available software (ACD/LC Simulator). A model that accounts for the changing structure and retention of proteins with temperature is proposed, delivering similar retention accuracy as found with simulation of small molecule separations.
Proteins and peptides are analytes of increasing interest within the pharmaceutical industry. While retention modelling has been successfully used for optimization of analytical scale separations of small molecules for approximately 30 years, it has not been expanded to accurate modelling of protein separations.
In this paper we describe the methods used to create retention models that allow accurate simulation of protein separations in commercially available software.
Experimental Conditions
Isocratic retention models were used in combination with numerical solutions to calculate retention times for gradient conditions (1).
Six proteins with MW ~25,000 Da were chromatographed and modelled by reversed phase chromatography (RPC) and ion exchange chromatography (IEC).
ACD/LC Simulator (2) was used for gradient modelling using the following equations:
RPC/HIC model lnk = a + b x + c x2 IEC/HILIC model lnk = d + e ln(x)
For small molecules, modelling gradient shape and temperature simultaneously has been found to be very effective, however, this was found to be insufficient for proteins and a second order temperature model was applied.
lnk = f + g / T + h / T2
As heat is applied a protein unfolds and refolds, and it is proposed that these changes lead to changes in protein retention with temperature.
Results
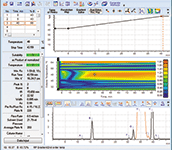
Combined gradient and temperature models were evaluated in RPC and IEC mode by fitting the models based in nine experiments (3 gradient times x 3 temperatures). Predictions were then made for four additional combinations of gradient time and temperature.
For a simple linear gradient, the accuracy of prediction was found to be systematic and greater than 98% for retention time (ΔtR <2%) and greater than 80% for peak width (Δw <20%). The impact of 20% peak width error is not significant for optimization purposes, and is similar for proteins and small molecules. Predictions for more complex, multi-step gradients showed similar accuracy.


Conclusions
RPC and IEC gradient chromatography, at different temperatures, can be modeled with the same accuracy for proteins as for small molecules. A second order temperature model is likely needed due to the unfolding of proteins at higher temperatures. Modeling of other types of separations, including HILIC and HIC as previously described by Snyder and colleagues (1), should also be possible.
References
(1) L.R. Snyder and J.W. Dolan, High-Performance Gradient Elution: The Practical Application of the Linear-Solvent-Strength Model (Wiley, Hoboken, NJ 2007).
(2) ACD/LC Simulator, Advanced Chemistry Development, Inc., Toronto, ON., Canada, www.acdlabs.com/lcsimulator (2017).


Advanced Chemistry Development, Inc.
8 King Street East, Suite 107, Toronto, Ontario, Canada M5C 1B5
tel. 1(800) 304-3988, email info@acdlabs.com.
Website: www.acdlabs.com
















SEC-MALS of Antibody Therapeutics—A Robust Method for In-Depth Sample Characterization
June 1st 2022Monoclonal antibodies (mAbs) are effective therapeutics for cancers, auto-immune diseases, viral infections, and other diseases. Recent developments in antibody therapeutics aim to add more specific binding regions (bi- and multi-specificity) to increase their effectiveness and/or to downsize the molecule to the specific binding regions (for example, scFv or Fab fragment) to achieve better penetration of the tissue. As the molecule gets more complex, the possible high and low molecular weight (H/LMW) impurities become more complex, too. In order to accurately analyze the various species, more advanced detection than ultraviolet (UV) is required to characterize a mAb sample.



