Rapid Multidimensional Liquid–Gas Chromatography for the Analysis of Saturated Hydrocarbon Contamination in Foods containing Vegetable Oil
LCGC Europe
A fast heart-cutting liquid chromatography–gas chromatography (LC–GC) method for the analysis of mineral oil saturated hydrocarbons (MOSHs) in a range of widely consumed foods is described.
This article describes a rapid heart-cutting liquid chromatography–gas chromatography (LC–GC) method for the analysis of mineral oil saturated hydrocarbons (MOSHs) found in a range of widely-consumed foods, including crisps, margarine, tinned tuna and vegetable oils.
The automated LC–GC experiments were performed using a system equipped with a syringe-type interface capable of both heart-cutting and comprehensive two-dimensional analysis. The first dimension separation was achieved on a silica column operated under isocratic conditions using hexane. The heart-cuts were then transferred to a programmed temperature vaporizer. After the large volume injection (LVI), the target analytes were rapidly separated (~ 9 min) using a micro-bore GC capillary column. The overall LC–GC run time enabled the analysis of around four samples in an hour. Various degrees of MOSH contamination — ranging from "not detected" to around 390 mg/kg — were discovered in the thirty samples that were subjected to analysis.
The contamination of foods with mineral oil saturated hydrocarbons (MOSHs) is an important issue. Mineral oil is derived from crude oil and mainly consists of MOSHs and mineral oil aromatic hydrocarbons (MOAHs). MOSHs are formed of straight and branched alkanes, as well as cyclic constituents such as naphthenes (1). Many papers based on this specific type of contamination have been reported over the last two decades. Grob et al. for example, reported various sources for this type of contamination, including mineral batching oil used for spinning jute, as well as lubricating oils and release agents used in the food industry (2,3). Printing inks that use mineral paraffins as solvents were found to have contamination levels between 10–100 mg/kg in foods contained in cardboard boxes (4). A case of mineral oil contamination in meats and eggs, caused by animal feed containing fat from waste collection sites, has also been reported (5). Another source of contamination is from the atmosphere. Neukom et al. investigated the presence of C20–C50 MOSHs in particulate matter from polluted air and suggested that this was an important source of MOSH contamination in vegetable oils (6).



In general, the presence of MOSHs in vegetable oils has been well-documented (6–9): Fiselier and Grob analysed sunflower oils using LC–GC analysis and discovered MOSH contamination levels between 2.7–32 mg/kg (7). It was suggested in this study that, apart from the atmosphere, contamination was also related to harvesting, storage, transport and processing. Two-hundred and twenty vegetable oil samples were analysed by Wagner et al., with levels of MOSH reaching 80 mg/kg (8). Recently, Tranchida et al. analysed several commercial vegetable oils and found some high levels of MOSH contamination, particularly for a pomace oil sample (9).
The use of food grade (white) mineral oil that contains only saturated hydrocarbons and no aromatics is allowed in the food industry.
The Joint FAO/WHO Expert Committee on Food Additives (JECFA) published a list of admissible daily intake (ADI) values for white mineral oils (10): for a high viscosity white oil the highest ADI value suggested was 20 mg/kg, while the lowest was 0.01 mg/kg for a low viscosity product. Recently, the European Food Safety Authority (ESFA) suggested an ADI value of 12 mg/kg bw (body weight)/day for high viscosity white mineral oils (11).
At the moment there are no legal concentration limits for MOSHs in foods, except for one specification defined by the EU Commission (50 mg/kg) for Ukrainian sunflower oil, following a critical case of contamination (12). What is known is that MOSHs are probably the most abundant contaminants in the human body, with the intake starting from the first day of our lives (13). Furthermore, the exact toxicological effects of such a high and constant exposure to MOSHs is still not known (14).
From an analytical perspective, heart-cutting LC–GC with a flame ionization detector (FID) is a suitable method for the determination of MOSHs in foods. Recently, Tranchida et al. developed a rapid, sensitive LC–GC method for the analysis of MOSHs in a selection of vegetable oils (9). The current research is based on the use of the fast LC–GC to analyse vegetable oils, as well as other commonly consumed foods, such as crisps, margarine and tinned tuna. Various degrees of contamination were found in the samples chosen.
Experimental
Samples and sample preparation: All the samples reported in Table 1 were purchased from supermarkets located in Messina, Italy, except for some of the extra virgin olive oils produced from a non-industrial source in the countryside near Messina (Table 1), kindly donated by staff from our laboratory. The vegetable oils were diluted to a final concentration of 25% v/v in hexane before LC–GC injection. The tinned tuna samples were filtered, and the vegetable oil collected and dried using Na2SO4. The vegetable oil was filtered again and then diluted to a final concentration of 25% v/v in hexane. The crisp samples (500 mg) were extracted with hexane (1 mL) for 1 h, then dried under a gentle nitrogen stream and diluted to a final concentration of 25% v/v in hexane. The margarine samples (500 mg) were extracted three times with hexane (1 mL each), then dried under a gentle nitrogen stream and diluted to a final concentration of 25% v/v in hexane.


Table 1: Samples subjected to LCâGC analysis and their contamination values.
LC–GC analysis: All two-dimensional LC–GC applications were performed on an LC×GC system (Shimadzu, Kyoto, Japan) consisting of a Shimadzu Prominence LC-20A system, equipped with a CBM-20A communication bus module, two LC-20AD dual-plunger parallel-flow pumps, a DGU20A online degasser, an SPD-20A UV detector, a CTO20A column oven and an SIL-20AC autosampler. Data were acquired by the LCsolution software (Shimadzu).
LC conditions: A 100 mm × 3 mm internal diameter (i.d.) × 5 µm dp silica column (Supelcosil LC-Si, Supelco, Milan, Italy) was operated under isocratic conditions, using hexane as a mobile phase (0.35 mL/min). The injection volume was 20 µL. At the end of the heart cut, the column was backflushed with CH2Cl2. A Shimadzu AOC-5000 autoinjector equipped with a dedicated dual side-port syringe was used as a transfer device (a detailed description will be given in the Results and Discussion section).
A Shimadzu GC2010 Plus, equipped with an Optic 3 injector (ATAS GC International, Eindhoven, The Netherlands). The Optic 3 injector was temperature-programmed as follows: from 75 °C (1 min) to 360 °C at 250 °C/min. The injection mode was split at a ratio of 200:1 for 1 min, then splitless for 1 min, then 50:1 for the remaining analysis time. Data were acquired by the GCsolution software (Shimadzu).
GC conditons: An SLB-5ms [silphenylene polymer, virtually equivalent in polarity to poly(5% diphenyl/95% methylsiloxane)] 15 m × 0.10 mm i.d. × 0.10 µm df column (Supelco) was heated from 50 °C (1 min) to 360 °C (4 min) at 70 °C/min. The hydrogen carrier gas was supplied at an initial pressure of 529 kPa (constant linear velocity was 100 cm/s). FID (360 °C) sampling frequency was 50 Hz.
Multidimensional LC×GC software (Shimadzu, Duisburg, Germany) enabled the control of each instrument through the respective native software. The software controlled the transfer process, which will be described in detail in the Results and Discussion section.
Results and Discussion
The multidimensional LC–GC system used in the present research can be used in either the heart-cutting or "comprehensive" mode. In the latter configuration, the entire initial sample is analysed in both dimensions. Chromatography band transfer is achieved through a modified 25-µL syringe. A schematic of the transfer device, in the cutting configuration, is illustrated in Figure 1. The lower part of the syringe barrel is linked, via two transfer lines, to the exit of the LC detector (line A) and to waste (line B). A Teflon plug is situated at the end of the syringe plunger, with the latter characterized by a lower outer diameter with respect to the i.d. of the barrel, enabling eluent flow inside the syringe. In the waste mode, the syringe plug is positioned below both lines and the effluent is directed to line B. In the cut position, the syringe plug is located between the two lines and the effluent flows through line A to the GC injector (the configuration represented in Figure 1).

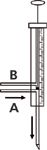
Figure 1: Schematic diagram of the syringe transfer device used for multidimensional LCâGC analysis in the cutting configuration.
In the present work, the first-dimension effluent was directed to waste until 1.5 min. From 1.5–2.0 min the LC effluent (the MOSH-containing fraction) was directed to the GC injector. Dedicated LC×GC software controlled the transfer process: after 1.5 min, the LC pumps were deactivated and the syringe was directed to the injector port. In the latter position, the plunger was moved to the cut position, and the LC pumps were activated for a time corresponding to the transfer period, namely, 30 s (175 µL). At the end of the transfer process the pumps were stopped, the plunger injected the remaining volume of LC-effluent and the fast GC analysis started. After each stop-flow period the gradient programme can be resumed, using exactly the same solvent composition as before the interruption. There is also a software window for the automatic calculation of the transfer line delay. This option is useful to determine the delay time of the LC fraction, from the UV detector to the injector port as a result of the transfer line volume.
The LC–GC method used, developed and validated in previous research is rapid and sensitive. The total analysis time is 14 min and the limit of quantification (LOQ) is 2 mg/kg (9). The first dimension is very useful because it frees the (possible) MOSH fraction from the lipids, which are retained on the silica phase. After transfer of the MOSH fraction, the primary column is backflushed with CH2Cl2. The scope of the present research was to apply the method not only to vegetable oils, but also to other foods which derive, or are prepared, using vegetable oil. Because MOSH contamination is also associated with industrial processing and transportation, a series of extra virgin olive oils, produced from a non-industrial source in the countryside near Messina, were evaluated.
Prior to food analysis, a blank sample (hexane) was subjected to LC–GC analysis to assess the possible contamination of the mobile phase and/or of the instrumentation. As can be seen in Figure 2, if contamination was present it was below the limit of detection (0.6 mg/kg) of the method. The impurities eluting between 3–4 mins were of no concern from an analytical perspective because they did not co-elute with the MOSH fraction.

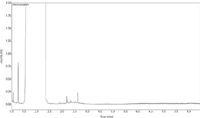
Figure 2: GC chromatogram for a blank sample (hexane).
In this study thirty food products were analysed consisting of four crisp samples, four margarine samples, five tinned tuna samples, and seventeen vegetable oil samples (which included fourteen extra virgin olive samples, one corn oil sample, one peanut oil sample and one sunflower sample).The results are listed in Table 1. All the values reported in the table are average results, derived from two consecutive LC–GC applications.
For the potato crisp samples no information was given on the packaging on the type of vegetable oil used. A 500mg quantity was extracted for 1 h with 1 mL of hexane. The second extraction gave a negligible response. The crisp contamination levels were rather limited, with all but one of the samples under the 4 mg/kg level (for sample 01 no contamination was detected). However, for this type of food, the sample subjected to hexane extraction is obviously formed mainly of potato, containing only a minimum quantity of vegetable oil. For this reason, crisp sample 04, which was characterized by a concentration of MOSH ≈ 20 mg/Kg was most probably produced using a rather highly contaminated oil. Therefore, no estimation can be made on the degree of contamination of that vegetable oil.
There was also no information on the type of vegetable oil used for the margarine samples. A 500-mg quantity was extracted for a 20 min period with 1 mL of hexane, three times consecutively. The fourth extraction gave a negligible response. Two of the margarine samples (01 and 02) were characterized by moderate contamination levels; whereas the other two samples presented a high degree of contamination (> 200 mg/kg), with one (03) nearing the 400 mg/kg mark. The amount of vegetable oil used in the production of the margarines was in the 60–70% range (reported on the container). Because such contamination levels are extremely high the following experiment was performed: 3 mL of hexane were dried under a nitrogen stream. The residue was dissolved in 1 mL of hexane and subjected to LC–GC analysis. Again, no response was observed.
Regarding the vegetable oil used in the production of tinned tuna — in this case olive oil — the degree of variability was high: sample 01 was characterized by a low contamination level (8.9 mg/kg), samples 03 and 04 by moderate ones and samples 02 and 05 by a contamination slightly exceeding 100 mg/kg and 80 mg/kg respectively. Commercial olive oil is technically a mixture of refined and extra virgin olive oil. A fast GC–FID chromatogram for tuna sample 05 is shown in Figure 3. As can be seen, the chromatogram is characterized by a typical MOSH "hump" centred at the C28 n-alkane in this case and contained in the C21–C35 range. Also highlighted in the figure are the predominant odd-numbered n-alkanes, which derive from the olive plant.

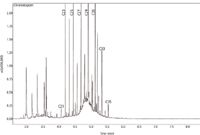
Figure 3: GC chromatogram for the tinned tuna sample 05 with a MOSH concentration of 83.7 mg/kg.
In this research five extra virgin olive oils (EVOOs), produced on family-run farms were also subjected to LC–GC analysis. For three of the EVOO samples the possible contamination was below the method's LOQ. The remaining two oils were characterized by low MOSH concentrations. It must be noted that the "family" oil group presented a 60% degree of negative response. It should be noted that no MOSHs were found in two out of nine commercial EVOO products; the remaining seven samples presented a low degree of contamination in the 2.5–10.5 mg/kg range, confirming the results obtained in previous research (9). It must also be added that in that study no EVOO samples were free from contamination in the four commercial products that were analysed. The production of EVOO essentially requires mechanical extraction only and the MOSH levels are, therefore, generally limited. The remaining three vegetable oils (corn oil, peanut oil and sunflower oil) contained moderate MOSH contamination. The production of seed oils is based on solvent extraction and a refining process and the possibility of MOSH contamination is, therefore, higher.
Conclusions
The results obtained in the present study indicate that the level of MOSHs present in vegetable oils, and in foods containing such oils, is strongly related to the number of industrial phases that an oil or an oil-containing food is subjected to. The "family-sourced" oils were characterized by the lowest degree of contamination. As expected, vegetable oil-containing foods that are rarely produced using EVOO were nearly always contaminated. The only exception was a crisp sample (01) and this was probably because this type of food contains a low amount of vegetable oil, and not because the oil used was free from MOSHs. The highest concentration of MOSHs found was 388.4 mg/kg for the margarine sample (03). Such a degree of contamination is certainly an excessive one, a statement which is also valid for the second most contaminated sample (margarine 04 = 220.4 mg/kg). Although the production of margarine requires a certain amount of industrial processing, there is no way to pinpoint where such a high level of contamination comes from.
The multidimensional LC–GC system is fairly new to this field and has been employed effectively and continuously during the last year. The instrumentation has also been used in the comprehensive two-dimensional mode. Currently, a rapid LC–GC method to analyse both MOSHs and MOAHs in foods is being developed. The presence of MOAHs in foods is certainly of greater concern from a toxicological perspective.
Peter Q. Tranchida is Associate Professor in Food Chemistry at the School of Pharmacy in Messina, Italy. He has co-authored approximately 80 publications and presented work in over 130 oral/poster presentations in national and international meetings. His main research activities are focused on the analysis of volatiles and semivolatiles contained in complex samples, by using advanced gas chromatographic methodologies. In particular, he has acquired considerable experience in the use of fast and very fast gas chromatographic techniques. He is actively engaged in the development and application of classical multidimensional GC, comprehensive two-dimensional GC (GC×GC) and LC–GC (LC×GC) systems.
Mariosimone Zoccali is currently a Ph.D. student in Food Chemistry and Safety at the University of Messina, Italy. His main research activities are focused on the development of heart-cutting and comprehensive multidimensional chromatography methods (GC–GC, LC–GC, GC×GC, LC×GC), combined with mass spectrometric detection, for food analysis.
Paola Dugo is Full Professor of Food Chemistry at the University of Messina, Italy, and at the "Campus Biomedico" in Rome. Her research interests include the development of comprehensive chromatography, and high throughput separation methods applied to the study of Citrus products (essential oils and juices),compounds with a possible biological activity in natural samples (carotenoids, anthocyanins and coumarins), plant essential oils, the aromatic fraction of wine and other alcoholic beverages, and of triglycerides in food lipids. Prof Dugo is the author of approximately 130 scientific papers, and has been invited to be a speaker at several national and international congresses.
Luigi Mondello is Full Professor of Analytical Chemistry at the Dipartimento Farmaco-chimico of the University of Messina, Italy, and at the University "Campus Biomedico" in Rome. He is the author of approximately 200 scientific papers and 29 book chapters. In addition, he is co-editor of a book on multidimensional chromatography (Wiley), and editor of a book on comprehensive chromatography in combination with mass spectrometry (Wiley).
His research interests include one-dimensional chromatography techniques, combined particularly with mass spectrometry, as well as multidimensional chromatography methods, such as LC–GC–MS, GC–GC– MS, GC×GC, LC×LC, LC×GC, and their use in the study of complex samples.
References
(1) V. Simanzhenkov, R. Idem, Crude Oil Chemistry, Marcel Dekker, New York, (2003).
(2) K. Grob, M. Lanfranchi, J. Egli and A. Artho, The Journal of AOAC International, 74(3), 506–512 (1991).
(3) K. Grob, A. Artho and M. Biedermann, J. Egli, Food Additives & Contaminants, 8(4), 437–446 (1991).
(4) C. Droz and K. Grob, Zeitschrift für Lebensmitteluntersuchung und -forschung, 205(3), 239–241 (1997).
(5) K. Grob, M. Vass, M. Biedermann and H.P. Neukom, Food Additives & Contaminants, 18(1), 1–10 (2001).
(6) H. P. Neukom, K. Grob, M. Biedermann and A. Noti, Atmospheric Environment, 36(30), 4839–4847 (2002).
(7) K. Fiselier and K. Grob, European Food Research and Technology, 229(4), 679–688 (2009).
(8) C. Wagner, H.P. Neukom, K. Grob, S. Moret, T. Populin, L.S. Conte, Mitt. Lebensm. Hyg., 92, 499–514 (2001).
(9) P.Q. Tranchida, M. Zoccali, G. Purcaro, S. Moret, L. Conte, M. Beccaria, P. Dugo and L. Mondello, Journal Chromatogr. A, 1218(42), 7476–7480 (2011).
(10) Summary of Evaluations Performed by the Joint FAO/WHO Expert Committee on Food Additives, April 25 (2003).
(11) Scientific Opinion on the Use of High Viscosity White Mineral Oils as a Food Additive, ESFA Journal 7, 1387 (2009).
(12) Summary Minutes of the Meeting of the Standing Committee on the Food Chain and Animal Health, June 20, (2008).
(13) N. Concin, G. Hofstetter, B. Plattner, C. Tomovski, K. Fiselier, K. Gerritzen, S. Fessler, G. Windbicher, A. Zeimet, H. Ulmer, H. Siegl, K. Rieger, H. Concin and K. Grob, Food and Chemical Toxicology, 46(2), 544–552 (2008).
(14) K. Grob, European Journal of Lipid Science and Technology, 110(11), 979–981 (2008).









New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.
 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")




