Rapid and Sensitive Determination of Residues of Triphenylmethane Dyes and Their Metabolites in Animal Tissues by LC–MS/MS
This method has greater sensitivity and a shorter LC analysis time than the AOAC method. It also requires less instrument maintenance, enhances column lifetime by requiring a smaller sample volume, and reduces cost by replacing acetonitrile with methanol in the mobile phase.
This article describes a fast, sensitive, selective, and robust method for determining triphenylmethane dyes and their metabolites in animal tissue samples by liquid chromatography with tandem mass spectrometry (LC–MS/MS). The separation of analytes from matrix-interfering components was performed using a C18 column with a mobile phase composition of water and methanol containing 0.1% formic acid and 5 mM ammonium formate as additives in both phases. To minimize sample matrix effects and analyte loss during sample preparation, extracted-matrix calibration in combination with isotope dilution was used for analyte quantification. There were two MS/MS transitions selected for each analyte for analyte confirmation, further enhancing the method’s selectivity and accuracy. The method was validated using salmon and chicken tissue samples.
Triphenylmethane dyes have been used as antimicrobial, antifungal, and antiparasitic agents to treat and prevent parasitic and fungal infections in aquaculture products, such as fish, for over 80 years because of their low cost and high efficiency against these infections (1). After usage, these dyes can be partially converted to their leuco-metabolites. These dyes and their metabolites can persist in edible fish tissues for several months (2). Because of concerns about their toxicity and mutagenicity, triphenylmethane dyes have been prohibited for treating fish used for human consumption by many countries, including the United States and European Union (EU) member states (3–5). To monitor these compounds in the food supply chains and safeguard the implementation of regulations, sensitive and selective analytical methods are required. In the EU, analytical methods used to determine these residues in aquaculture products must be sensitive enough to meet the European minimum required performance limit (MRPL) of 2 μg/kg for the sum of malachite green (MG) and leucomalachite green (LMG) (6). In the United States, the minimum sensitivity of 1 μg/kg was established by the U.S. Food and Drug Administration for regulatory testing (7). No MRPL was set for other dyes. Recently, several liquid chromatography with tandem mass spectrometry (LC–MS/MS) methods have been developed for the analysis of dyes and their metabolites (8–13). Hurtaud-Pessel and his colleagues developed a method for the identification and quantification of the triphenylmethane dyes, malachite green (MG), crystal violet (CV), brilliant green (BG), leuco-malachite green (LMG), and leuco-crystal violet (LCV) in trout tissue (8). The quantification of the analytes was carried out by an extracted-matrix calibration method. This method was granted as the Association of Official Agricultural Chemists (AOAC) First Action Method for seafood analysis (9). This method was also validated for other animal species by Andersen (10) and investigated further in an AOAC collaborative study in 2015 (11).
In this study, a rapid, sensitive, and selective LC–MS/MS method was developed based on a simplified AOAC method (12). Sample matrix effects and analyte loss during sample preparation can be compensated for by using extracted-matrix calibration in combination with isotope dilution. The method has been validated using salmon fish and chicken tissue sample matrices. Compared to the AOAC method, this new method has higher sensitivity and a shorter LC analysis time. In addition, the method needs less instrument maintenance and enhances column lifetime because of the smaller sample volume injected. Because this method uses less expensive methanol to replace acetonitrile as a mobile phase, it can reduce cost for routine testing laboratories.
Experimental
Chemicals and Materials
Analytical standards and isotope-labeled internal standards were obtained from Sigma-Aldrich: malachite green oxalate salt; leuco-malachite green; crystal violet; leuco-crystal violet; brilliant green; malachite green-d5 picrate; leuco-malachite green-d5; crystal violet-d6 trihydrate; and leucocrystal violet-d6. The chemical structures of triphenylmethane dyes and their metabolites were illustrated in references (6,7). LC–MS grade solvents methanol, acetonitrile, and water, as well as other chemicals such as formic acid, ammonium formate, hexanes, magnesium sulfate (anhydrous), hydroxylamine hydrochloride, and ascorbic acid, were also obtained from Sigma-Aldrich. Disposable syringes (1 mL), and syringe filters (0.22 μm) were obtained from VWR. Polypropylene centrifuge tubes (15 mL and 50 mL), and amber color autosampler vials and caps were obtained from PerkinElmer Inc. The endcapped C18 sorbent was obtained from UCT. Organic salmon fish, chicken tissue, and other seafood samples were obtained from local stores (Toronto, Ontario, Canada).
Standard Preparation
Stock solutions of each standard and internal standard at 1 mg/mL were prepared in acetonitrile by weighing accurately 5 mg of each standard (calculating the content of the active substances), and dissolving each standard in 5 mL of acetonitrile. Secondary stock solutions of each standard at 10 μg/mL were prepared in acetonitrile by appropriate dilution of their corresponding stock solutions. These solutions were stored in a freezer at −20 °C in amber vials (protecting them from light) and were found stable for at least three months. A mixed intermediate standard solution at 1 μg/mL (containing MG, LMG, CV, LCV, and BG) and a mixed intermediate internal standard solution at 1 μg/mL (containing MG-d5, LMG-d5, CV-d6, and LCV-d6) were prepared by diluting the secondary stock solutions in acetonitrile, respectively. These solutions were stored in a freezer at −20 °C in amber glass vials and were found stable for at least 1 month. These intermediate solutions were diluted in acetonitrile to prepare nine levels of working standard mixed solutions (WS1 to WS9) at concentrations of 0, 1, 2, 5, 10, 20, 40, 50, and 100 ng/mL (corresponding to 0, 0.05, 0.1, 0.25, 0.5, 1.0, 2.0, 2.5, and 5.0 μg/kg in samples) and a working internal standard mixed solution (WS-IS) at 40 ng/mL. These solutions were prepared fresh daily.
For the AOAC method, a hydroxylamine solution (9.5 g/L) in water and an ascorbic acid solution (1g/L) were prepared by the procedures described in AOAC method (9). Reconstitution solution was prepared by combining 1 mL of ascorbic acid solution (1 g/L) with 100 mL acetonitrile and mixing well.
Sample Preparation
Animal tissue samples were homogenized and ground with dry ice in a food processor to obtain uniformed powder and kept at –20 °C overnight to allow sublimation of residual dry ice and then stored at –20 °C until analysis. Certified organic chicken and salmon samples were used as the blank matrix for method validation.
Extracted Matrix Calibrant Samples
For this study, 2.00 g (±0.02 g) of the homogenized sample was weighed into each of nine 50 mL disposable centrifuge tubes. Each of the nine samples was spiked with a 100 μL internal standard working solution (WS-IS) and then fortified with 100 μL aliquots of WS1, WS2, WS3, WS4, WS5, WS6, WS7, WS8, and WS9, respectively. The extracted matrix calibrant samples were thus fortified with 0, 0.05, 0.1, 0.25, 0.5, 1.0, 2.0, 2.5, and 5.0 μg/kg of analytes and 2.0 μg/kg of internal standards. Readers can refer to AOAC method (9) for more detailed procedures. The calibrants were allowed to equilibrate for 15 min while being protected from light before extraction.
Note that the AOAC method specifies that five extracted matrix calibrants are prepared in the range 0 to 2 μg/kg with concentrations 0, 0.5, 1.0, 1.5, and 2.0 μg/kg of analytes (9). In this study, the range was extended from 0 to 5 μg/kg to ensure that residues found in samples would fall within the calibration range.
Sample Extraction
Two grams (±0.02 g) of the homogenized sample was weighed into a 50 mL disposable centrifuge tube. Each sample was spiked with 100 μL internal standard working solution (WS-IS) and then allowed to equilibrate for 15 min while being protected from light before extraction. Then, 8 mL of acidified acetonitrile (1% formic acid) and 1.0 g anhydrous magnesium sulfate was added to each tube. Each sample was vortex mixed for 1 min and shaken (15 min) using a multitube vortexer. Samples were then centrifuged for 8 min at 4 °C and 4000 rpm. The supernatant was transferred to a clean 15-mL centrifuge tube and evaporated to dryness at 50 °C with N2 gas. The extracted matrix calibrant samples and test samples were reconstituted with 0.8 mL of acidified acetonitrile (0.1% formic acid) and vortexed for 1 min.
The sample solutions were chilled at −20 °C for 0.5 hour, and then centrifuged for 6 min at 4 °C and 4000 rpm. The solution was filtered through a 0.22 μm filter into an autosampler vial for LC–MS/MS analysis.
Quality Control
A laboratory reagent blank (LRB) was prepared and tested first to ensure that there was no interference or contamination from reagents or materials used and from the sample preparation processes. All sample blanks (organic chicken tissue and salmon fish) were tested as control matrix blanks to check for analyte peaks or interfering components. Sample matrix effects (ME%) were evaluated by comparing the responses between a post-extract spiked sample and a sample in a neat solution. Sample extraction efficiencies (EE%) or analyte recoveries from sample matrix were calculated by comparing the response of a fortified sample (spiked before extraction) to the response of post-extract spiked sample. Because of sample matrix effects and analyte loss during sample preparation, analyte quantification in the sample was carried out by extracted-matrix calibration in combination with isotope dilution method.
Analytical Conditions
Analyte separation was performed using a ultrahigh-pressure liquid chromatography (UHPLC) system (PerkinElmer LX-50) with an Epic C18 column (3.0 μm, 100 × 2.1 mm) and a mobile phase composition of water and methanol with 0.1% formic acid and 5 mM ammonium formate as additives in both phases (flow rate at 0.25 mL/min). Then, 5.0 μL of sample was injected with a partial loop injection mode, and the total LC running time was 10.5 min. Analyte detection was achieved using a triple quadrupole mass detector (PerkinElmer QSight 420) under positive electrospray ionization (ESI) mode. Multiple multiple reaction monitoring (MRM) transitions were monitored for each compound to evaluate potential interfering components for any transitions in the sample matrices. The ion source conditions were ESI voltage, 5000 V; drying gas, 120 L/h; nebulizer gas, 220 L/h; source temperature, 350 oC; and HSID temperature, 220 oC.
Results and Discussion
LC–MS/MS Method Optimization
For mass detection, multiple MS/MS transitions for each analyte were evaluated in this study to improve analyte confirmation and method’s accuracy. The optimized MRM parameters for the final selected quantifier and qualifier ions were listed in Table I. To separate analytes from matrix components, reversed-phase HPLC with C18 column was used in this study. All five analytes can be separated within 5 min, and the total run time (including column clean and equilibration time) is 10.5 min, which is much shorter than the time (20 min) required by the AOAC method (9). Because of the high sensitivity of the mass detector, a smaller sample injection volume (5 μL) is required in this method compared to 20 μL in the AOAC method, which can increase column lifetime and reduce instrument maintenance. In addition, methanol was used as an organic mobile phase component in this study instead of acetonitrile as in the AOAC method, further reducing the cost for routine testing laboratories.


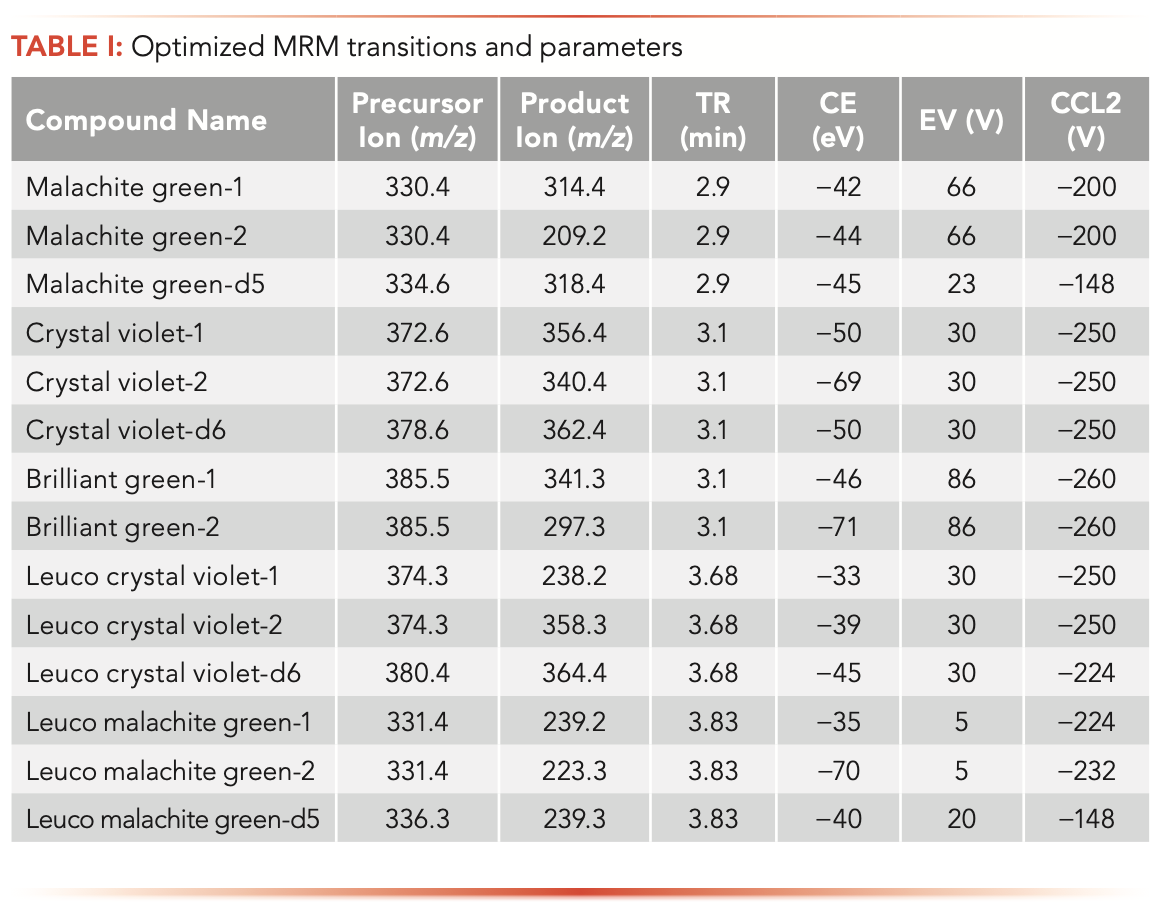
Sample Preparation
Two sample preparation methods were reported in literature (9,12). One is the AOAC method (9). In this method, hydroxylamine was used during sample extraction to prevent the conversion of MG to LMG, and ascorbic acid was added in the reconstitution solution to avoid the transformation of MG to carbinol and prevent demethylation reactions. Another method is a simplified AOAC procedure done by Kaplan’s team (12). They used acidified acetonitrile (1% acetic acid) for extraction and acidified acetonitrile (0.1% acetic acid) for the reconstitution solution because using acid can prevent the transformation of MG to carbinol as well as demethylation reactions. In this study, the two sample preparation methods were initially compared, and no significant difference in the results was found; therefore, the simplified method was used for further method validation.
Sample Matrix Effects and Analyte Recovery
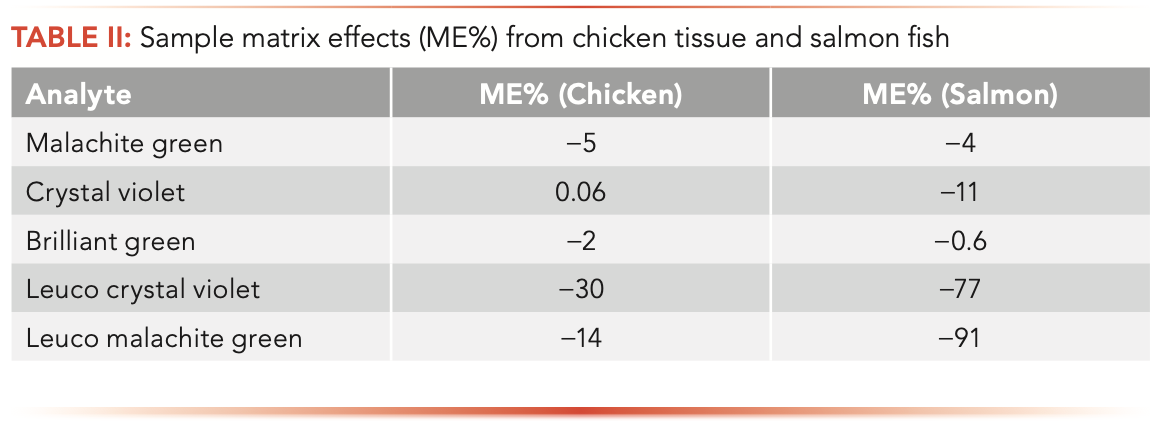
One of the biggest challenges in LC–MS/MS method development and validation is sample matrix effects because they can affect data quality and method’s selectivity, sensitivity, and accuracy, especially for complex sample matrices such as various food and animal tissue samples. In this study, ME% were evaluated by comparing the responses of analytes in a post-extract spiked sample with those in a spiked neat solution, as shown in the following equation:


Positive values indicate signal enhancement, whereas negative values indicate signal suppression effects. As shown in Table II, most analytes suffered from signal suppression effects in the studied sample matrices. To reduce sample matrix effects, two different sample cleanup steps were evaluated: liquid–liquid extraction (LLE) with hexanes, and d-SPE with endcapped C18 sorbent. However, no significant improvement in matrix effects was observed; thus, these clean-up steps were not used in the final method.

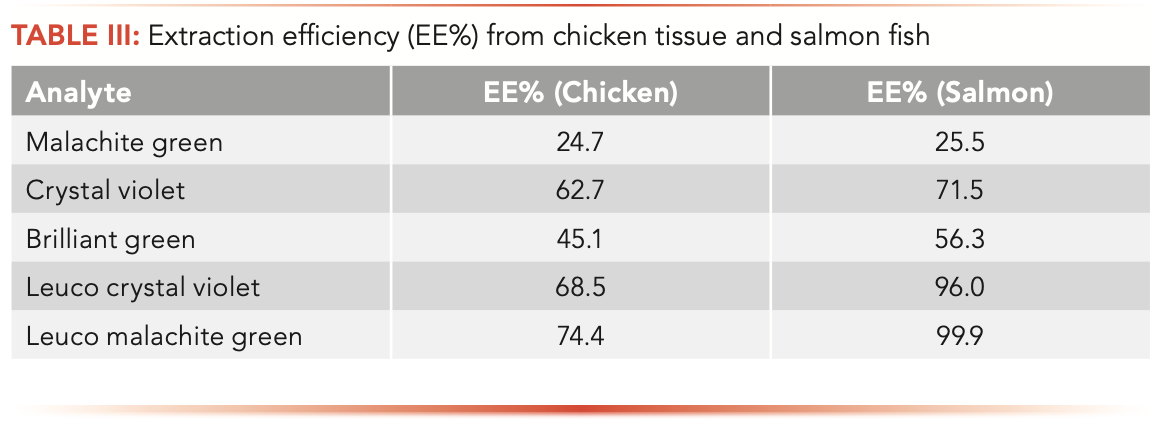
Analyte recoveries, or EE%, from the sample matrix were calculated by comparing the response of a fortified sample (spiked before extraction) to the response of post-extract spiked sample, as shown in the following equation:


As shown in Table III, for most analytes in the studied sample matrices, analyte recoveries are ranging from 60–99%, demonstrating adequate extraction efficiency of the method. Because of sample matrix effects and extraction deficiency (<80%) for some analytes, isotope dilution assay or extracted-matrix calibration method (fortifying analytes in each sample matrix with different concentrations before sample extraction) was recommended for analyte quantification.

Method Performance and Validation
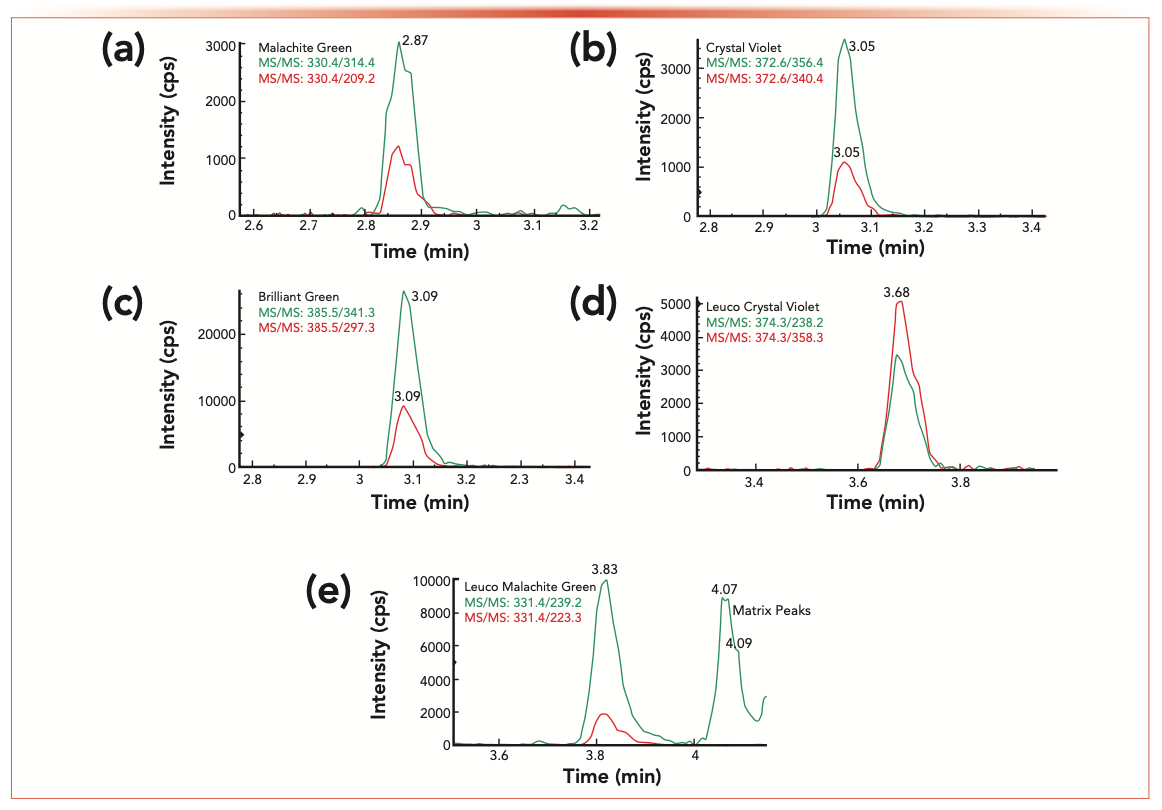
Nine concentration levels of calibration standards were prepared in both a neat solution (solvent-only calibration) and in each sample matrix (extracted matrix calibration). All the calibration curves show good linearity with the correlation coefficients (R2) greater than 0.99. The limit of quantification (LOQ) of the method was estimated by the signal-to-noise ratio (S/N = 10) of each analyte peak (quantifier) in the spiked sample matrix. Overall, the LOQ values of the methods for all analytes are below 0.1 μg/kg for the studied sample matrices, demonstrating good method sensitivity. The method’s selectivity and analyte confirmation from samples can be evaluated by comparing the analyte retention time and mass spectrum information (such as the peak area ratios of qualifier to quantifier ions of the analyte) between reference standard and samples. According to regulatory guidance on analytical method validation, at least two structurally specific MS/MS transitions should be used in a LC–MS/MS method (14–16). For example, the overlapped two MS/MS chromatograms of the five analytes in a spiked salmon sample were illustrated in Figure 1. No interference or contamination from reagents, glassware, and sample tubes was observed in this study (no analyte was detected in any of the LRB samples). No analytes were found above the LOQ of the method from the studied sample matrix blanks. Method precision was assessed based on replicate analyses of spiked samples (five replicates). The precision was then calculated based on the coefficient of variation (RSD%) of the collected data. The RSDs were less than 15% for all the analytes in the spiked samples. Method precision or reproducibility could also be evaluated by the linearity of the extracted matrix calibration curves because each level of calibration samples was prepared by fortifying analytes to samples prior to sample extraction and then going through the whole sample preparation and analysis procedures. Method accuracy assesses how close the experimental value is to the expected value and can be evaluated by the relative recovery of a known amount of analyte spiked to a sample matrix (LFM samples). In this study, the relative recovery was calculated based on extracted-matrix calibration method in combination with isotope dilution to compensate for sample matrix effects and sample loss (low extraction efficiency for some analytes, as shown in Table III) during sample preparation. The accuracy of the method is good for all the analytes from the spiked LFM samples as shown in Table IV, with relative recoveries ranging from 82.9 to 120%.

FIGURE 1: (a–e) Two overlapped MS/MS chromatograms for each of the five analytes: (a) malachite green; (b) crystal violet; (c) brilliant green; (d) leuco crystal violet; and (e) leuco malachite green in salmon tissue spiked at 0.5 μg/kg for each analyte. For the two MS/MS transitions of each analyte, the quantifier MS/MS (green) and qualifier MS/MS (red) are shown.

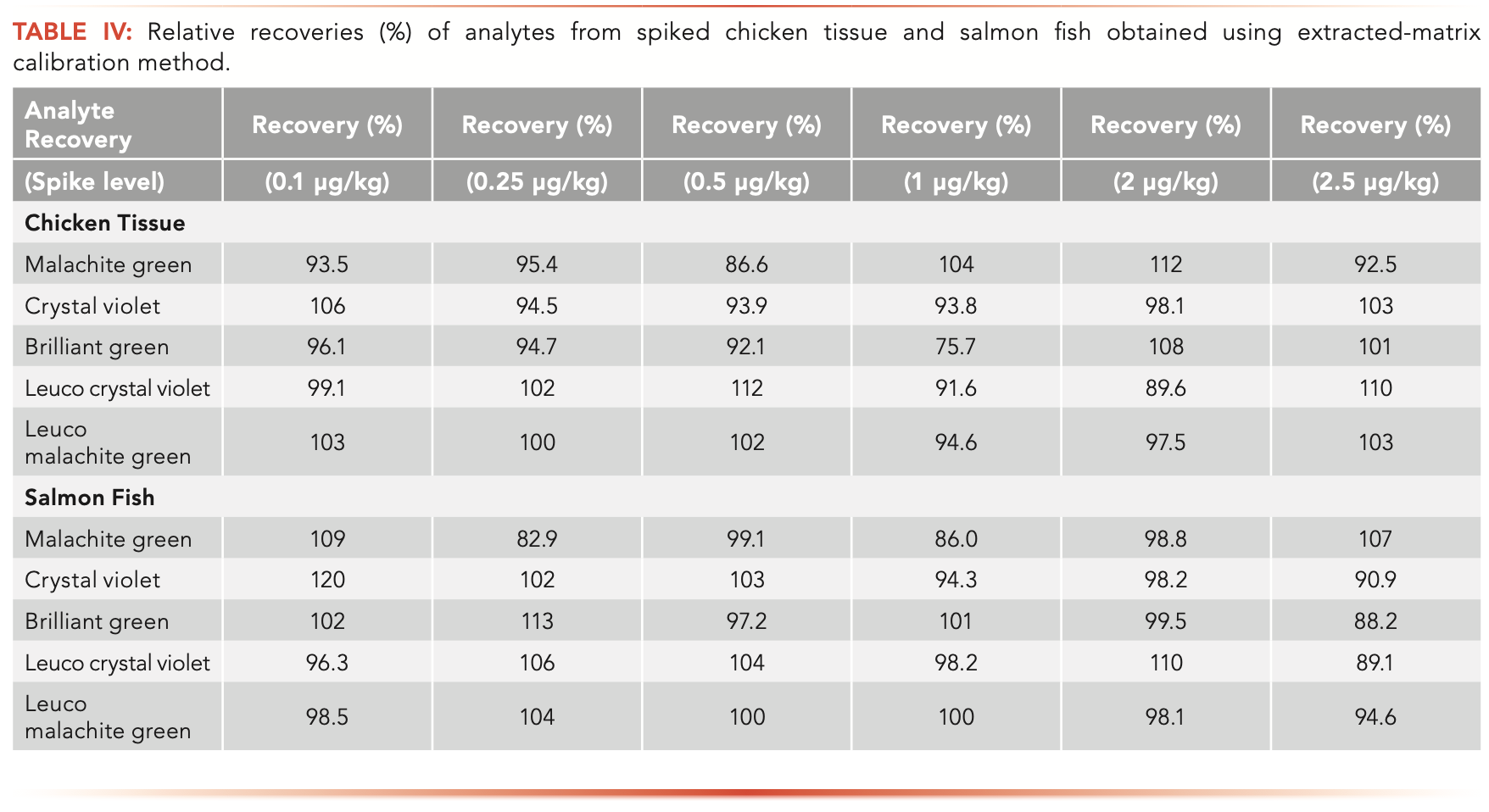
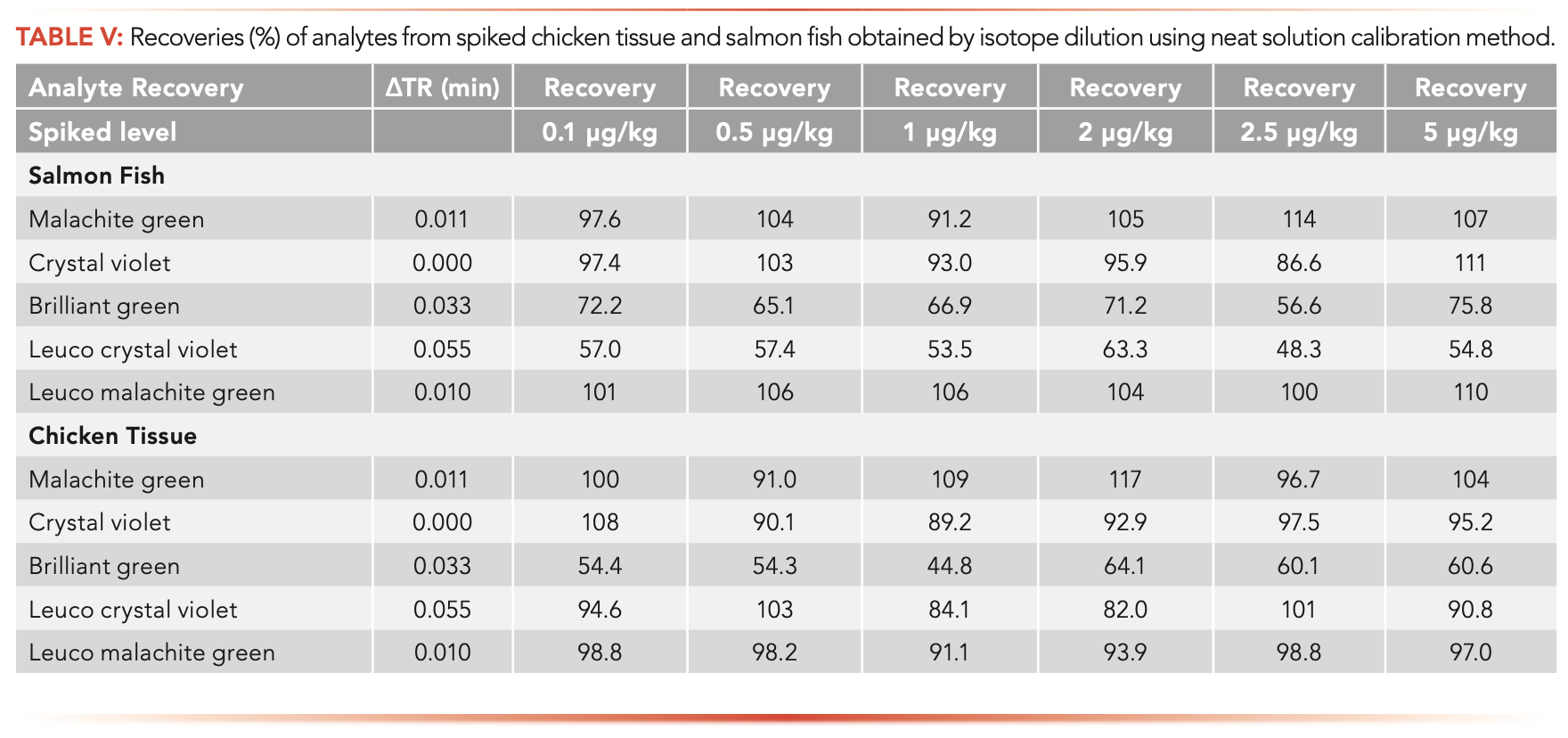
It should be noted that stable isotope dilution assay (SIDA) couple with LC–MS/MS method can overcome sample matrix effects and variations in sample preparation and analysis, and thus has been widely used for accurate quantification of chemicals, such as mycotoxins in various sample matrices using a simple calibration prepared in neat solution (17). However, in this study, we found that the accuracy was not good (<80%) for the determination of analyte brilliant green in all sample matrices studied, because isotope-labeled brilliant green is not available commercially and crystal violet-d6 was used as its internal standard for brilliant green quantification. More interestingly, we found that the accuracy for analysis of leuco-crystal violet was also not good in some sample matrices (such as in salmon fish) as demonstrated in Table V, although its isotope labeled internal standard was used in this study. This is possibly because of the so-called “isotope effect” (IE), which can cause larger retention time difference (ΔTR) between the analyte and its isotope analogue, and thus lead to different matrix effects for the analyte and its internal standard (18). Therefore, sample matrix effects could not be corrected by internal standard calibrations prepared in neat solution. Thus, the best quantification method for this analyte is the extracted-matrix calibration method as demonstrated in this method.

Method robustness was studied by slightly changing the experimental parameters such as the mobile phase gradients and HPLC columns from a different batch. No significant differences in method’s performance and analyte results were observed and thus confirmed the robustness of the method.
The method was applied to the determination of these compounds in various sea food products, no analytes were found above the limits of quantification of the method.
Conclusion
This study demonstrates a fast, sensitive, selective, and robust method for the analysis of triphenylmethane dyes and their metabolites in animal tissue samples by LC–MS/MS. In comparison with the AOAC action method, this new method provides several advantages including higher sensitivity, shorter LC analysis time, smaller sample injection volume (which leads to longer column lifetime and less instrument maintenance), and reduced cost for routine testing laboratories by using less expensive methanol to replace acetonitrile as a mobile phase. Using extracted-matrix calibration in combination with isotope dilution in the method, sample matrix effects and any analyte loss during sample preparation can be effectively corrected. The method was validated using chicken tissue and salmon fish sample matrices spiked at several concentration levels and was applied for analysis of these analytes in animal tissue samples with good linearity, precision and accuracy.
References
(1) Alderman, D. J. Malachite Green: A Review. J. Fish Dis. 1985, 8 (3), 289–298. DOI: 10.1111/j.1365-2761.1985.tb00945.x
(2) Chan, D.; Tarbin, J. A.; Stubbings, G.; Kay, J.; Sharman, M. Analysis of Incurred Crystal Violet in Atlantic Salmon (Salmo salar L.): Comparison Between the Analysis of Crystal Violet as an Individual Parent and Leucocrystal Violet and as Total Crystal Violet after Oxidation with 2,3-dichloro-5,6-dicyanobenzoquinone. Food Addit. Contam. A 2012, 29 (1), 66–72. DOI: 10.1080/19440049.2011.623283
(3) Wan, H.; Weng, S.; Liang, L.; Lu, Q.; He, J. Evaluation of the Developmental Toxicity of Leucomalachite Green Administered Orally to Rats. Food Chem. Toxicol. 2011, 49 (12), 3031–3037. DOI: 10.1016/j.fct.2011.10.003
(4) Culp, S. J.; Mellick, P. W.; Trotter, R. W.; Greenlees, K. J.; Kodell, R. L.; Beland, F. A. Carcinogenicity of Malachite Green Chloride and Leucomalachite Green in B6C3F1 Mice and F344 Rats. Food Chem. Toxicol. 2006, 44 (8), 1204–1212. DOI: 10.1016/j.fct.2006.01.016
(5) Srivastava, S.; Sinha, R.; Roy, D. Toxicological Effects of Malachite Green. Aquatic Toxicol. 2004, 66 (3), 319–329. DOI: 10.1016/j.aquatox.2003.09.008
(6) Commission Decision 2004/25/EC of 23 December 2003, Amending Decision 2002/657/EC as Regards the Setting of Minimum Required Performance Limits (MRPLs) for Certain Residues in Food of Animal Origin. Off. J. Eur. Communities, 2004, L6, 38–39. https://www.legislation.gov.uk/eudn/2004/25/pdfs/eudn_20040025_adopted_en.pdf
(7) Chen, G.; Miao, S. HPLC Determination and MS Confirmation of Malachite Green, Gentian Violet, and Their Leuco Metabolite Residues in Channel Catfish Muscle. J. Agric. Food Chem. 2010, 58 (12), 7109–7114. DOI: 10.1021/jf9043925
(8) Hurtaud-Pessel, D.; Couëdor, P.; Verdon, E. Liquid Chromatography–Tandem Mass Spectrometry Method for the Determination of Dye Residues in Aquaculture Products: Development and Validation. J. Chromatogr. A 2011, 1218 (12), 1632–1645. DOI: 10.1016/j.chroma.2011.01.061
(9) Hurtaud-Pessel, D.; Couëdor, P.; Verdon, E. Determination of Residues of Three Triphenylmethane Dyes and Their Metabolites (Malachite Green, Leuco Malachite Green, Crystal Violet, Leuco Crystal Violet, and Brilliant Green) in Aquaculture Products by LC/MS/MS: First Action 2012.25. J. AOAC Int. 2013, 96 (5), 1152–1157. DOI: 10.5740/jaoacint.13-142
(10) Andersen, W. C.; Casey, C. R.; Nickel, T. J.; Young, S. L.; Turnipseed, S. B. Dye Residue Analysis in Raw and Processed Aquaculture Products: Matrix Extension of AOAC INTERNATIONAL Official Method 2012.25. J. AOAC Int. 2018, 101 (6), 1927–1939. DOI: 10.5740/jaoacint.18-0015
(11) Schneider, M. J.; Andersen, W. C. Determination of Triphenylmethane Dyes and Their Metabolites in Salmon, Catfish, and Shrimp by LC–MS/MS Using AOAC First Action Method 2012.25: Collaborative Study. J. AOAC Int. 2015, 98 (3), 658–670. DOI: 10.5740/jaoacint.14-263
(12) Kaplan, M.; Olgun, E. O.; Karaoglu. O. A Rapid and Simple Method for Simultaneous Determination of Triphenylmethane Dye Residues in Rainbow Trouts by Liquid Chromatography–Tandem Mass Spectrometry. J Chromatogr A. 2014, 1349 (4), 37–43. DOI: 10.1016/j.chroma.2014.04.091
(13) Eich, J.; Bohm, D. A.; Holzkamp, D.; Mankertz, J. Validation of a Method for the Determination of Triphenylmethane Dyes in Trout and Shrimp with Superior Extraction Efficiency. Food Addit. Contam. Part A 2020, 37 (1), 84–93. DOI: 10.1080/19440049.2019.1671611
(14) US Food and Drug Administration, Bioanalytical Method Validation Guidance for Industry 2018, 83 (99), 23690 (Docke No. FDA-2013-D-1020). 1–41. https://www.fda.gov/media/70858/download
(15) European Commission, 2002/657/EC: Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results 2002, L221, 8–36. https://op.europa.eu/en/publication-detail/-/publication/ed928116-a955- 4a84-b10a-cf7a82bad858%20
(16) European Commission, Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed. SANTE/ 12682/2019. https://www.eurlpesticides.eu/userfiles/file/EurlALL/AqcGuidance_ SANTE_2019_12682.pdf
(17) Wu, J.; Ordinario, T. Analysis of 12 Regulated Mycotoxins in 8 Food Matrices by Single Immunoaffinity Cleanup and Isotope Dilution LC–MS/MS. LCGC North Am. 2021, 39 (s1), 21–27. https://www.chromatographyonline.com/view/analysis-of-12-regulated-mycotoxins-in-8-food-matrices-by-single-immunoaffinity-cleanup-and-isotope-dilution-lc-ms-ms
(18) Rychlik, M.; Asam. S. Stable Isotope Dilution Assays in Mycotoxin Analysis. Anal. Bioanal. Chem. 2008, 390 (2), 617–628. DOI: 10.1007/s00216-007-1717-x
Jingcun Wu is with PerkinElmer Health Sciences Canada, Inc., in Bolton, Ontario, Canada. Direct correspondence to: jingcun.wu2@perkinelmer.com



 leaves, green essence or organic extract in glass bottles. Natural ingridients use for alternative medicine, healthy cosmetics concept. | Image Credit: © Elena - stock.adobe.com")











Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.

Characterizing Polyamides Using Reversed-Phase Liquid Chromatography
May 5th 2025Polyamides can be difficult to characterize, despite their use in various aspects of everyday life. Vrije Universiteit Amsterdam researchers hoped to address this using a reversed-phase liquid chromatography (RPLC)-based approach.



New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.
, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

