Rapid and Direct Quantitation of Pharmaceutical Drugs from Urine Using MALDI-MS
Special Issues
An application of MALDI-MS in qualitative and quantitative analysis of pharmaceutical compounds spiked in urine is demonstrated.
The quantitative estimation of drugs from biofluids in pharmacokinetic studies requires high-throughput bioanalytical methods. We report a rapid, simple, and efficient method for simultaneous quantitative determination of different pharmaceutical drugs directly from urine samples. Acetaminophen, griseofulvin, ampicillin, and verapamil spiked in human urine were characterized directly by matrix-assisted laser desorption–ionization time-of-flight mass spectrometry (MALDI-TOF-MS) followed by tandem MS to confirm their identities. Furthermore, quantitative profiling of drugs was carried out directly from spiked urine samples using MALDI-TOF-MS without any chromatographic separation. Linearity was observed for the relevant concentration ranges of the metabolites found in urine. The quantitative analysis successfully produced acceptable precision and high accuracy of recoveries of drugs.
Pharmacokinetics plays a very important role in the drug development process for prioritizing lead compounds. It involves the investigation of absorption, distribution, metabolism and excretion (ADME) of drugs from the body, and helps in screening of drug candidates. After a drug is administered into the blood stream orally or intravenously, it is distributed to the site of action and metabolized or biotransformed. Finally, the drug and its metabolites are excreted from the body through the kidneys in the form of urine. Slow or incomplete excretion can lead to the accumulation of higher concentrations of the drug at the site of action. Quantitative estimation of the excreted drug is important to determine the therapeutic dosage of the drug, its toxicological effect, the rate of excretion, and the amount of drug absorbed by the body. Simultaneous analysis and quantitation of different drugs with diversity in their chemical nature, especially from biological fluids, is indeed a challenging task.
Mass spectrometry (MS) has established itself as a crucial bioanalytical tool in the pharmaceutical industry and the drug development process (1,2). Versatility coupled with high sensitivity, resolution, and dynamic range make MS a method of choice in pharmacokinetic studies. Liquid chromatography–mass spectrometry (LC–MS)-based methods for quantitative determination have been used routinely in the past decade and are now considered benchmarks for bioanalytical analysis (3–6). However, chromatographic techniques require a lot of attention and can be time consuming during analytical method development. In drug development processes where time is a critical factor, any time-consuming methods are undesirable. Often, there are also large sample sets in the analytical pipeline that demand high-throughput analytical techniques (7).
Direct mass analysis and quantitation is an efficient alternative that is not yet being used to its optimum potential. This is primarily because of concerns about introducing an impure sample without chromatographic separation. Major time-consuming steps and tedious sample preparation can be avoided by introducing the sample directly into the ionization source of a mass spectrometer (8). However, most of the recently developed direct mass analytical techniques that can bypass the chromatographic step have not yet been fully explored for quantitative analysis (9,10). Among the direct mass analytical methods, matrix-assisted laser desorption–ionization mass spectrometry (MALDI-MS) has several advantages. MALDI-MS systems are robust, require minimal sample preparation, are tolerant to salts and buffers, and offer high-throughput analysis (11–15). Furthermore, the generation of mainly singly charged ions with high resolution provides uncomplicated spectra (16). The interference of matrix ions in small molecule regions of the mass spectra can be offset by choosing the matrices carefully and using MS-MS validation of the precursor ions. MALDI-MS has been demonstrated for both qualitative and quantitative analysis of targeted low-molecular-weight compounds, pharmaceuticals, and other small-molecule metabolites (13,17–24). Particularly, the use of MALDI-MS in imaging of drugs distributed in cells has also gained much interest (25–27). Considering the limited time span for bioanalytical study during drug development processes, MALDI-MS is an alternative for drug screening from biological fluids because of its inherent advantages.
Urine has been routinely used as a biofluid for analytical studies because of its noninvasive sampling, lower protein content, and larger quantities that can be collected for multiple cycles of analysis. Urine excreted from the human body also contains a higher concentration of endogenous and exogenous metabolites. In this study, we explored direct and quantitative analysis of several drugs spiked in blank urine samples using MALDI-MS. A simple and rapid quantitative workflow is demonstrated for the analysis of drugs from human urine without any further sample preparation, along with their characterization with comprehensive collision-induced fragmentation.
Materials and Methods
Chemicals
Acetaminophen (N-(4-hydroxyphenyl)acetamide), griseofulvin ((2S,5'R)-7-chloro-3',4,6-trimethoxy-5'-methylspiro[1-benzofuran-2,4'-cyclohex-2-ene]-1',3-dione), ampicillin ((2S,5R,6R)-6-[[(2R)-2-amino-2-phenylacetyl]amino]-3, 3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid), verapamil (2-(3,4-dimethoxyphenyl)-5-[2-(3,4-dimethoxyphenyl)ethyl-methylamino]-2-propan-2-yl-pentanenitrile), 2,4-diamino-6-(2-fluorophenyl)-1,3,5-triazine, 2,5-dihydroxybenzoic acid, trifluoroacetic acid, methanol, and acetonitrile were purchased from Sigma-Aldrich. Deionized water with a specific resistivity of 18.2 MΩ was obtained using an SG ultrapure water unit.
Sample Preparation
A 100 mM stock solution of each drug was prepared in methanol, diluted to the desired concentration, and mixed in equal volume. The final concentration of each drug molecule in the mixture was as follows: 2 mM acetaminophen, 40 µM griseofulvin, 1 mM ampicillin, and 2 µM verapamil. 2,4-Diamino-6-(2-fluorophenyl)-1,3,5-triazine was used as an internal standard for quantitative analysis. A 1000 ppm stock solution of internal standard was prepared in 1:1 (v/v) methanol–water and diluted to 10 ppm. 2,5-Dihydroxybenzoic acid (DHB) was used as the MALDI matrix. A 20-mg/mL DHB solution was prepared in 1:1 (v/v) acetonitrile–0.1% trifluoroacetic acid. Later a mixture of DHB and internal standard was prepared for quantitative MALDI-MS.
Urine was collected from a healthy volunteer and immediately aliquoted and stored at -20 °C. To prepare urine samples spiked with pharmaceutical drugs, 100 µL of urine was mixed with 100 µL of the drug mixture and centrifuged at 4 °C for 1 h at 13,200 rpm. The supernatant was collected in a separate vial. A series of calibrants was prepared from urine samples by spiking varying concentrations of the drug mixture.
Mass Spectrometry
A Synapt MALDI-TOF-MS system (Waters) was used in the positive ion mode for all of the MS experiments. Before the experiments, the instrument was properly tuned and calibrated for mass accuracies with standards and protocols as prescribed. Detector voltage and quadrupole settings were adjusted to obtain maximum sensitivity. The laser energy (Nd:YAG laser, 355 nm) was optimized to achieve a good signal-to-noise ratio above the threshold energy of the MALDI matrix.
All of the samples were analyzed on standard 96-well stainless steel MALDI target plates that were precleaned thoroughly. Before spotting, the samples were premixed with MALDI matrix (a mixture of DHB and internal standard). The samples (1 µL per spot) were spotted manually in replicates using a micropipette. The quantitative data were acquired by using an instrument in automatic acquisition mode. During automatic acquisition, the instrumental conditions and acquisition time were kept uniform across the MALDI target plate. For detection and identification of drug molecules, precursor ions of each drug molecule were subjected to collision-induced dissociation (CID) fragmentation.
Data Analysis and Method Validation
The qualitative data analysis was performed using Masslynx 4.1 (Waters) and mMass (open source) software. For quantitative data analysis, an in-house developed software, "MQ," was used to generate calibration curves based on ratios of peak intensities of analyte to internal standard. These calibration curves were used to determine the concentration of drugs in quality control samples and drug-spiked urine samples. Six replicates were used for each calibration point and the average was calculated for generating the calibration curve. A linear regression equation was used in calculating the correlation coefficient (R2 ), slope, intercept, recoveries, and relative standard deviations (RSD).
Results and Discussion
Qualitative Determination of Drug Molecules
In this study, commonly used pharmaceutical drugs were selected from different classes of drugs according to their function, structure, and molecular weight (Figure 1).


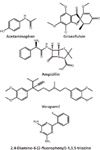
Figure 1: Chemical structures of pharmaceutical drugs spiked in urine and the internal standard used in this work.
Acetaminophen is a frequently used analgesic that has the lowest molecular weight among the drugs selected for this study. Griseofulvin is a metabolic product of P. griseofulvum, and it has been used as an antifungal agent to treat dermatophytic infections. Ampicillin has been used extensively as an antibiotic in bacterial infections. It is a -lactam antibiotic and belongs to the penicillin family. Verapamil, a derivative of phenylalkylamine, is an effective antiarrhythmic and vasodilator. It is also used as a calcium channel blocker. The qualitative study of drug molecules was carried out in two stages. First, MALDI-MS of the drug mixture premixed with the DHB matrix was performed (data not shown). All the drug molecules showed mainly protonated adduct of the respective molecular ion [M+H]+ and had good mass accuracy (within 10 ppm). DHB matrix peaks were also present in the spectra, but the drug molecule peaks were clearly resolved. Subsequently, the precursor ions of drugs were allowed to fragment in the collision chamber. The fragmentation patterns matched with those reported in previously published data and, thus, corroborated the identification of each drug molecule (28–31).


Figure 2: MALDI-MS spectra of pharmaceutical drugs spiked in urine and analyzed directly. 2,5-Dihydroxybenzoic acid (DHB) was used as matrix.
MALDI-MS was subsequently used directly on urine samples spiked with drugs followed by MS-MS validation as described above for the pure standards. Figure 2 shows the representative MALDI-MS of drugs from spiked urine samples. A zoomed-in portion of the spectra with a protonated and sodiated adduct of drug molecules is also shown in the figure. As expected, a very complex spectrum was obtained from spiked urine with many peaks seen across the entire mass range until m/z 600. This complexity can be attributed to the presence of various endogenous metabolites along with several possible adducts and fragments thereof. Accurate masses for protonated and sodiated adducts of all the spiked drugs were detected with good resolution and signal-to-noise ratio. MS-MS from precursor ions reveals the identity of analyte molecules unambiguously and is especially useful for analysis from complex samples such as urine. Table I summarizes the CID fragments of each drug molecule in MALDI MS-MS. The overall identification of drugs spiked in urine was performed reproducibly in a rapid manner. It is noteworthy that all the drugs were simultaneously detected directly from the urine without any chromatographic separation or sample preprocessing.

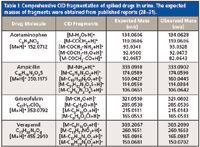
Table I: Comprehensive CID fragmentation of spiked drugs in urine. The expected masses of fragments were obtained from published reports (28â31).
Quantitative Determination of Drug Molecules
The quantitative trends of the drug molecules were studied by constructing calibration curves from both pure standards and subsequently from spiked urine samples. Calibration responses were measured as peak height ratios of the sample normalized against an internal standard. The internal standard compensates for the variation in signal response that arises because of inhomogeneity in sample spotting and fluctuation in instrumental parameters over time. The selection of the internal standard was based on the reproducibility of signals, miscibility, and noninterference with the drug (18,32). Isotopically labeled internal standards have been a preferred choice for users, but have disadvantages because of their cost and nonavailability. Nonlabeled standards provide an alternative to isotopically labeled internal standards. Previously published reports have successfully shown the usage of chemically nonanalogous compounds as internal standards (22,24). A symmetric triazine derivative was used as an internal standard, which is nonendogenous to urine metabolites. Table II represents the quantitative statistics of drug molecules directly quantified from a mixture of drugs. All drug molecules showed good linearity with correlation coefficients (R2) above 0.97. The calibration curve was prepared by taking average values of six replicates per dilution and the concentration range was started above the limit of quantitation (LOQ). The detection limit of standard solution for each drug molecule was estimated before quantitative analysis by taking those concentrations that showed a signal-to-noise ratio with peaks above 3. Using these criteria, all the drug molecules were obtained with detection limits at the low picomoles per microliter level. To verify the method, quality control samples containing drugs in low, mid, and high concentration ranges of calibration curves were analyzed on the same target plate. Further recoveries of drugs from quality control (QC) samples were calculated from the linear equation of calibration curve. All the drug molecules showed reproducible recoveries within 15% RSD in most cases, and extending up to 20% in a few, validating the linear relationship between concentrations of drugs and response from MALDI-MS data.

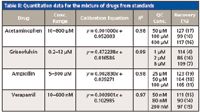
Table II: Quantitation data for the mixture of drugs from standards
The quantitative analysis of drugs was further extended to samples present in urine to demonstrate the utility of the method for biological fluids. The blank urine was fortified with drugs, which simulated the presence of a drug in excreted urine after the administration of a drug in the human body. Acetaminophen, griseofulvin, ampicillin, and verapamil were spiked in urine according to previously published reports (Table III) (33–36). Serial dilutions of the drugs were added to urine samples with no further dilution. The limit of detection (LOD) and LOQ of drug molecules were determined from spiked urine. Acetaminophen and ampicillin were detected at 7.6 and 8.7-ng/µL, whereas the detection limits for griseofulvin and verapamil were found to be 0.35 and 0.036 ng/µL. The LOD and LOQ were higher for the urine spikes when compared with the pure standards. Calibration curves were generated by plotting the concentration of drugs spiked and their ratios of peak height with internal standard. Table IV summarizes concentration ranges, linearity equations, correlation coefficients, and recoveries from spiked urine samples analyzed directly using MALDI-MS with no sample preprocessing. Excellent linearity was observed for each drug molecule with high correlation coefficients and reproducibility fulfilling the Food and Drug Administration's criteria of 20% RSD at LOQ and 15% elsewhere in the calibration range. The recoveries from QC samples were calculated as the mean of 12 samples. The recoveries of 91–112% were within 15% RSD, except for ampicillin, for which one of the QC samples had an RSD of 19%.


Table III: Concentration range used in quantitation of drugs from urine samples (33â36)
Once again, it is noteworthy that the entire analysis, starting from sample preparation to data analysis, was performed within a few hours and did not necessitate any chromatographic separation. The analysis time can further be reduced using automated spotting. Additionally, accuracy and reproducibility of data can also be improved with automation.
Until recently, MALDI was not considered a suitable ionization method for small-molecule analysis (<m/z 500) because of matrix interference in the low-molecular-weight region. However, several recent publications have demonstrated the utility of MALDI-MS in targeted small molecule analysis (22–24). Newer generations of MALDI-MS systems are equipped with high duty cycle analyzers and advanced electronics, which provide high resolution data and, subsequently, mass accuracies less than 10 ppm. An accurate identification of a molecule can be obtained based on peaks with high mass accuracy and subsequent CID fragmentation. Peak area–based quantitation can be challenging for metabolites and other small molecules from biological fluids in the absence of a chromatographic separation, because the contiguous peaks in the mass spectrum often are poorly resolved. Peak intensity–based quantitation demonstrated using large sample sets has previously shown efficiency comparable to that of peak area–based quantitation (22,24), and can be advantageous. This approach is quite useful for MALDI-MS, in which inconsistent desorption–ionization processes may lead to poor quantitation.

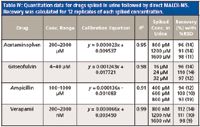
Table IV: Quantitation data for drugs spiked in urine followed by direct MALDI-MS. Recovery was calculated for 12 replicates of each spiked concentration.
Conclusions
The application of MALDI-MS in qualitative and quantitative analysis of pharmaceutical drugs spiked in urine was demonstrated. Drug molecules were characterized by MALDI-MS followed by CID fragmentation. High resolution and mass accuracies within 10 ppm along with MS-MS fragmentation patterns corresponding to the analytes of interest aid in unambiguous detection from urine. Peak height–based quantitation performed using a chemically unrelated internal standard on both pure standards and spiked urine samples showed excellent sensitivity, linearity, accuracy, and reproducibility. The method was validated with quality control checks. The method reported establishes the versatility of MALDI-MS in direct analysis from urine with minimal sample processing and without any chromatographic separation. Tolerance to sample impurities along with the higher throughput obtained using MALDI-MS can increase the efficiency of ADME and similar bioanalytical studies.
Acknowledgments
This research was funded by CSIR grants and the Department of Biotechnology (India) young investigator award (RGYI Grant 102/IFD/SAN/PR-2062/2009–2012). V.P. acknowledges funding from DBT Ramalingaswamy fellowship.
References
(1) E. Want et al., Spectrosc. Int. J. 17(4), 681–691 (2003).
(2) G. Hopfgartner and E. Bourgogne, Mass Spectrom. Rev. 22(3), 195–214 (2003).
(3) T.A. Gillespie and B.E. Winger, Mass Spectrom. Rev. 30(3), 479–490 (2011).
(4) A. Tolonen et al., Drug Discovery Today 14(3–4), 120–133 (2009).
(5) R.S. Plumb et al., Rapid Commun. Mass Spectrom. 22(14), 2139–2152 (2008).
(6) M.S. Lee and E.H. Kerns, Mass Spectrom. Rev. 18(3–4), 187–279 (1999).
(7) W.Z. Shou and J. Zhang, Expert Opin. Drug Metab. Toxicol. 6(3), 321–336 (2010).
(8) S.X. Yu et al., Anal. Chem. 81(1), 193–202 (2009).
(9) E. Chernetsova and G. Morlock, Bioanalytical Reviews 3(1), 1–9 (2011).
(10) E.S. Chernetsova and G.E. Morlock, Mass Spectrom. Rev. 30(5), 875–883 (2010).
(11) M. Bucknall et al., J. Am. Soc. Mass Spectrom. 13(9), 1015–1027 (2002).
(12) L.H. Cohen and A.I. Gusev, Anal. Bioanal. Chem. 373(7), 571–586 (2002).
(13) Z.-M. Masoud et al., Rapid Commun. Mass Spectrom. 18(2), 141–148 (2004).
(14) M.W. Duncan et al., Briefings Funct. Genomics Proteomics 7(5), 355–370 (2008).
(15) E. Szajli et al., Mol. Cell. Proteomics 7(12), 2410–2418 (2008).
(16) M. Karas et al., J. Mass Spectrom. 35(1), 1–12 (2000).
(17) P.J. Lee et al., Anal. Chem. 76(16), 4888–4893 (2004).
(18) S. Lekha and A.V. Dietrich, Rapid Commun. Mass Spectrom. 19(14), 1928–1936 (2005).
(19) S. Notari et al., J. Chromatogr., B 833(1), 109-116 (2006).
(20) J. A. Campbell et al., Anal. Lett. 40(16–18), 3107–3118 (2007).
(21) S. Notari et al., J. Chromatogr., B 863(2), 249–257 (2008).
(22) P. Markus and K. Michael, Rapid Commun. Mass Spectrom. 23(22), 3555–3562 (2009).
(23) D. Yukihira et al., Anal. Chem. 82(10), 4278–4282 (2010).
(24) A. Singh and V. Panchagnula, Anal. Methods 3(10), 2360–2366 (2011).
(25) D. Bonnel et al., Bioanalysis 3(12), 1399–1406 (2011).
(26) N. Takai et al., Rapid Commun. Mass Spectrom. 26(13), 1549–1556 (2012).
(27) J.D. Pallua et al., Current Trends in Mass Spectrometry (supplement to LCGC North Am.) 21–28 (October 2011).
(28) J. Borlak et al., Xenobiotica 33(6), 655–676 (2003).
(29) W. Weinmann et al., J. Mass Spectrom. 36(9), 1013–1023 (2001).
(30) C.R. Huang et al., J. Pharm. Biomed. Anal. 59, 157–161 (2012).
(31) H.N. Mistri et al., J. Chromatogr., B 850(1–2), 318–326 (2007).
(32) W.R. Wilkinson et al., Fresenius' J. Anal. Chem. 357(3), 241–248 (1997).
(33) E.-S.A. Ibrahim et al., Anal. Lett. 21(3), 423–434 (1988).
(34) H. Zia et al., J. Chromatogr. 181(1), 77–84 (1980).
(35) S. Heitmeier and G. Blaschke, J. Chromatogr., B 721(1), 93–108 (1999).
(36) E.A. Kasim et al., J. Pharm. Biomed. Anal. 30(4), 921–929 (2002).
Ajeet Singh, Nivedita Bhattacharya, Avinash Ghanate, and Venkateswarlu Panchagnula work in the Chemical Engineering Division at the National Chemical Laboratory in Pune, India. Direct correspondence to: v.panchagnula@ncl.res.in
















New TRC Facility Accelerates Innovation and Delivery
April 25th 2025We’ve expanded our capabilities with a state-of-the-art, 200,000 sq ft TRC facility in Toronto, completed in 2024 and staffed by over 100 PhD- and MSc-level scientists. This investment enables the development of more innovative compounds, a broader catalogue and custom offering, and streamlined operations for faster delivery. • Our extensive range of over 100,000 high-quality research chemicals—including APIs, metabolites, and impurities in both native and stable isotope-labelled forms—provides essential tools for uncovering molecular disease mechanisms and exploring new opportunities for therapeutic intervention.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.

