Native Separation-Mass Spectrometry in Biopharmaceutical Analysis
Analytical methods that allow separation and identification of therapeutic proteins under native conditions play a crucial role in studying their higher-order structures and structure–function relationships. Recently, hyphenated techniques that combine native-mode separation with native mass spectrometry (nMS) have emerged as highly valuable tools for the targeted assessment of these quality attributes. This article outlines current native separation strategies coupled with nMS designed to characterize biopharmaceuticals close to their natural state. The methods provide worthwhile insights into aspects like aggregation, charge variants, conjugate stoichiometry, affinity, and conformation. As multidimensional chromatographic techniques and ion-mobility spectrometry become more accessible in laboratories, further advances in the development of native hyphenated techniques capable of simultaneously providing compositional, structural, and functional information on biopharmaceuticals can be expected.
Over the past few decades, there has been substantial progress in the development of mass spectrometry-based tools for the structural analysis of intact proteins, driven by requirements in the fields of biopharmaceuticals, structural biology, and top-down proteomics. Proteins, as entities, frequently exhibit considerable heterogeneity reflecting a plethora of subtle structural modifications.
The characterization of these different proteoforms is a challenging but frequently indispensable task. For example, during development and production of therapeutic proteins, it is crucial to monitor several critical quality attributes (CQAs) that are linked to the presence of protein variants (1). In addition to chemical variations, factors such as conformational and supramolecular structures (including aggregates, conformers, and complexes), as well as the level of bioactivity, can contribute to the heterogeneity of biopharmaceuticals. Evaluating these properties requires the preservation of the natural conformation and functional state of the proteins of interest throughout the analysis process.
Native mass spectrometry (nMS) has been demonstrated to be a highly useful approach for gaining deeper understanding of the structure and behavior of proteins in conditions closely resembling their natural or native state (2). In this context, native reflects a protein’s biological status prior to ionization. This involves the protein being immersed in an aqueous solution with a pH and ionic strength conducive to preserving its natural folded conformation (3). nMS provides accurate mass determinations of intact proteins by employing electrospray ionization (ESI) that, in principle, can preserve conformation and non-covalent interactions of proteins in the gas phase (4). Consequently, nMS allows for the analysis of tertiary and quaternary structures, protein–substrate and protein–protein complexes, related stoichiometries and binding affinities, and even large macromolecular assemblies. When proteins are sprayed under native conditions, for example, using 100 mM ammonium acetate (AmAc) at pH 7 as solvent, they display charge state distributions (CSDs) that are distinctly different from those obtained under conventional, denaturing conditions:
a reduced number of charge states, resulting in considerably narrower CSDs, and a decrease in overall charge, shifting CSDs to the higher m/z region. The nature of a protein’s CSD can indicate whether, and to what extent, a protein is in its native conformation.
To obtain high-quality and information- rich protein mass spectra with nMS, it is necessary to work with purified samples (5). Extensive sample pretreatment is frequently required, particularly when the proteins of interest are part of complex matrices/samples. Furthermore, it is crucial to exchange nonvolatile buffers and salts in the sample for volatile substances, commonly AmAc, to ensure MS compatibility. As a result, the necessary sample preparation is often time-consuming and labor-intensive, with the added risk of inducing protein modifications, degradation, or denaturation. The burden on sample preparation can
be alleviated by incorporating efficient analytical separation prior to nMS detection. Hyphenation of protein separation techniques to nMS can strongly reduce the complexity of obtained mass spectra, facilitating the detection of low abundant protein species or proteoforms with subtle mass differences, up to the distinction of isomeric species. Additionally, analytical separations intrinsically encompass sample cleanup and the possibility of buffer exchange, circumventing ion suppression and unwanted adduct formations. Upfront separation helps filter out protein species formed as artefacts during the ionization process. For example, although intrinsically a soft ionization method, ESI can induce protein aggregate formation, particularly at high protein concentrations. Coupling size-based separation to nMS allows to differentiate between aggregates that genuinely exist in the sample and those that are merely false aggregates generated by the ESI process.
When the goal is to characterize higher-order structures or protein affinity by nMS, the hyphenated separation methods should preserve natural protein conformations and interactions. This requirement a priori excludes the commonly applied protein separation techniques reversed-phase liquid chromatography (RPLC) and hydrophilic interaction chromatography (HILIC), which by default use denaturing organic solvents in their eluents. When conducted aptly, which means using aqueous mobile phases of proper pH, ionic strength and temperature, separation techniques such as size-exclusion chromatography (SEC), ion-exchange chromatography (IEC), hydrophobic interaction chromatography (HIC), affinity chromatography (AC), and asymmetrical flow field flow fractionation (AF4) can, in principle, offer valuable protein resolutions while maintaining native conditions (6,7). In addition, electromigration-based techniques such as capillary zone electrophoresis (CZE), capillary isoelectric focusing (CIEF), and affinity capillary electrophoresis (ACE) can be conducted under native conditions achieving high separation efficiencies (8). Nevertheless, when directly coupled with nMS, adjustments to routinely used methods are necessary, as these often employ phosphate buffers or salt gradients that are not compatible with MS (9).
The objective of this paper is to provide a concise overview of the available online combinations of native separation and nMS, and how they can yield distinctive and exceptionally valuable information regarding a range of crucial properties in biopharmaceutical analysis. The topics covered are characterization of aggregates, charge variants, conjugates, affinity, and conformation of protein therapeutics (Figure 1). Each subject is introduced followed by typical examples that demonstrate the performance, applicability, and scope of the respective native techniques in more detail. More comprehensive overviews on the hyphenation of native protein separations with MS can be found elsewhere (6,8,10).


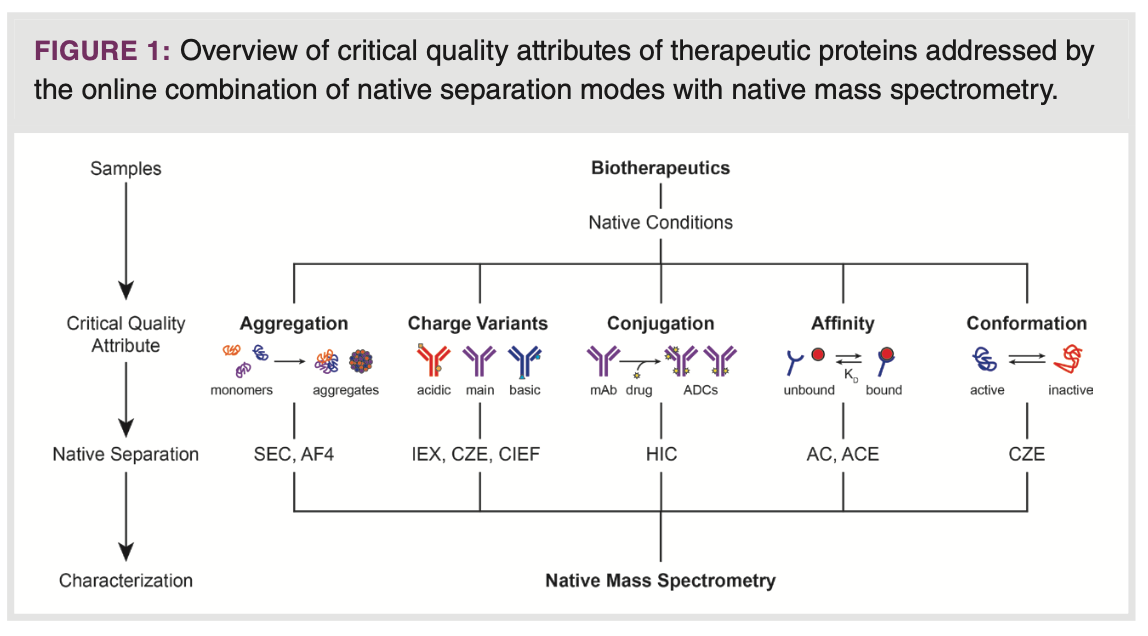
Protein Aggregates
Characterization of the size variants of therapeutic proteins is essential
to ensure the quality and safety of biopharmaceutical products. The presence of protein fragments and subunits is indicative for bioprocess performance and protein stability, whereas protein aggregates, such as dimers, can be highly immunogenic. Size-based separation techniques, such as SEC and AF4, can resolve proteins from their aggregates and fragments under native elution conditions. These circumstances are critical as most aggregates are held together by non-covalent interactions, which may be particularly sensitive to denaturing environments. The coupling of SEC and AF4 to nMS facilitates the identification of size variants while also providing information on other structural aspects, including post-translational modifications (PTMs).
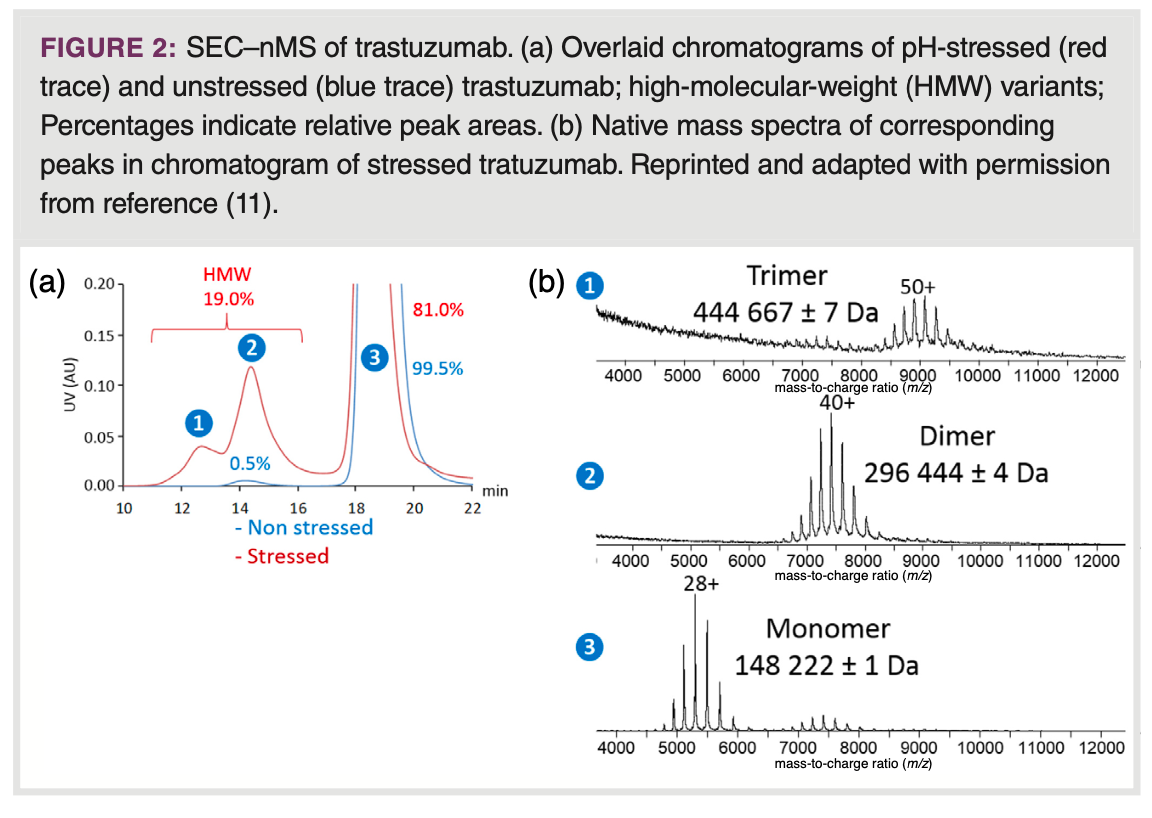
SEC–nMS: Native SEC for biopharmaceutical analysis is commonly carried out using aqueous eluents containing non-volatile buffer salts
at substantial concentrations. When coupling SEC to nMS, it becomes necessary to substitute these for volatile, MS-compatible, electrolytes. Using a mobile phase containing 100 mM AmAc (pH 6.8), SEC–nMS has demonstrated its ability to both identify and quantify side- and degradation products of pharmaceutical monoclonal antibodies (mAbs) (Figure 2) (11). Along similar lines, Haberger et al. used SEC–nMS to profile bispecific antibody samples for product-related impurities derived from incorrect light or heavy chain association during production, and aggregates, formed during storage and exposure to elevated temperature (12). Employing short columns of less than 5 cm filled with narrow-pore particles, SEC has been integrated with nMS to enable rapid and automated desalting of samples, without attaining protein separation (13). This concept has been utilized in online two-dimensional (2D)-LC schemes to combine native protein separations employing non-volatile salts with nMS. Fractions from MS-incompatible SEC modes were comprehensively analyzed by native short-column SEC using volatile eluents, allowing structural assessment of mAb size variants when coupled to nMS (9). In SEC employing long columns (15–30 cm) filled with wide-pore particles, the use of volatile mobile phases can result in compromised SEC performance, for example, distorted peaks and shifts in retention time due to unintended interactions with the SEC column and the stationary phase material. These interactions may also cause partial protein unfolding. Ventouri et al. demonstrated that when it comes to retaining native protein conformations in long-column SEC–nMS, AmAc is the most effective among options such as ammonium formate and bicarbonate, however, only when used at appreciable ionic strength (200 mM) (14). Such concentrations can still induce protein ionization suppression and in combination with common SEC flow rates requires relatively harsh ESI conditions, which potentially disrupt protein higher-order structures. This limitation can be alleviated, at least to some extent, by employing innovative bioinert SEC columns featuring chemically modified surfaces, which reduce unwanted protein interactions. This is illustrated by Murisier and colleagues in their work on size-variant analysis of various mAbs through SEC–nMS, using an eluent containing 50 mM ammonium acetate (15). An alternative solution lies in adopting micro-flow SEC–nMS, using 1.0 mm i.d. columns operated at 15 μL/min. This approach significantly improves ionization efficiency allowing use of concentrations as high as 400 mM AmAc to prevent protein interactions with conventional SEC stationary phases (16). The effectiveness of this approach was demonstrated by the analysis of fragments and aggregates
of trastuzumab and the tetrameric anticancer drug asparaginase (ASNase).

AF4–nMS: AF4 is a separation technique suitable for resolving large (bio)macromolecules, aggregates, and particles based on their size (17). The underlying separation mechanism relies on differential diffusion of analytes in a flow field perpendicular to the laminar flow of a carrier liquid in a thin open channel. Protein samples can be fractionated by size without the use of a stationary phase, excluding any adverse protein interactions with packing material. This allows for the use of MS-compatible mobile phases comprising relatively low concentrations (10–20 mM) of AmAc without losing separation performance. In addition, AF4 circumvents the potential physical stress exerted on protein structures while passing through the narrow channels of columns packed with stationary phase particles (18). These attributes are particularly valuable for the quality and stability assessment of relatively labile biopharmaceuticals, such as ASNase, where the tetramer serves as the actual active pharmaceutical compound. The AF4–nMS analysis of an unstressed ASNase sample revealed the presence of both octamer and monomer species next to the drug substance (19). It also unveiled the gas-phase dissociation of tetramer into monomer and association of tetramer to octamer during ESI, which would go undetected if direct nMS without prior separation was used.
Protein Charge Variants
Presence of charge variants is one of the key CQAs of biopharmaceuticals,
in particular for mAbs (20). The typical approach for characterizing protein charge profiles involves techniques such as ion exchange chromatography (IEX), capillary zone electrophoresis (CZE), and imaging capillary isoelectric focusing (iCIEF) in combination with UV-Vis absorbance detection. While these methods are efficient in a quality control setting, they do not allow unambiguous structural assignment of expected and unexpected peaks. Coupling with MS, however, represents a challenge, mainly due to the common use of non-volatile buffer or mobile phase components to achieve optimal separation performance. In recent years, native-based separation techniques in combination with nMS, have emerged as a solution for charge variant analysis (CVA). There are two primary advantages associated with using nMS for CVA. Firstly, there are fewer interfering ions in the higher mass-to-charge ratio (m/z) region. Secondly, proteoforms carrying different modifications are better resolved due to the wider m/z spacing among proteoforms at lower charge states.
IEX–nMS: Strong cation exchange chromatography (SCX) is a technique that separates protein species based on their surface charge. In general, SCX separation can be carried out under native conditions (21). Yan et al. outlined the coupling of SCX with nMS, employing a combined pH (5.6–7.4) and salt (20–150 mM) gradient for the CVA of various mAbs spanning a wide pI range. This approach allowed for the detection of minor modifications such as a non-consensus Fab glycosylation site (<0.1%) in the NIST mAb reference standard. Haberger and colleagues developed a SCX-MS method using a pH gradient ranging from 6.0 to 10.7 for the CVA of a 200 kDa T-cell bispecific humanized mAb, containing an additional Fab domain. With this approach, they not only identified a new N-glycosylation motif in the heavy chain CDR region, but also separated glycoforms carrying varying degrees of sialylation (22).
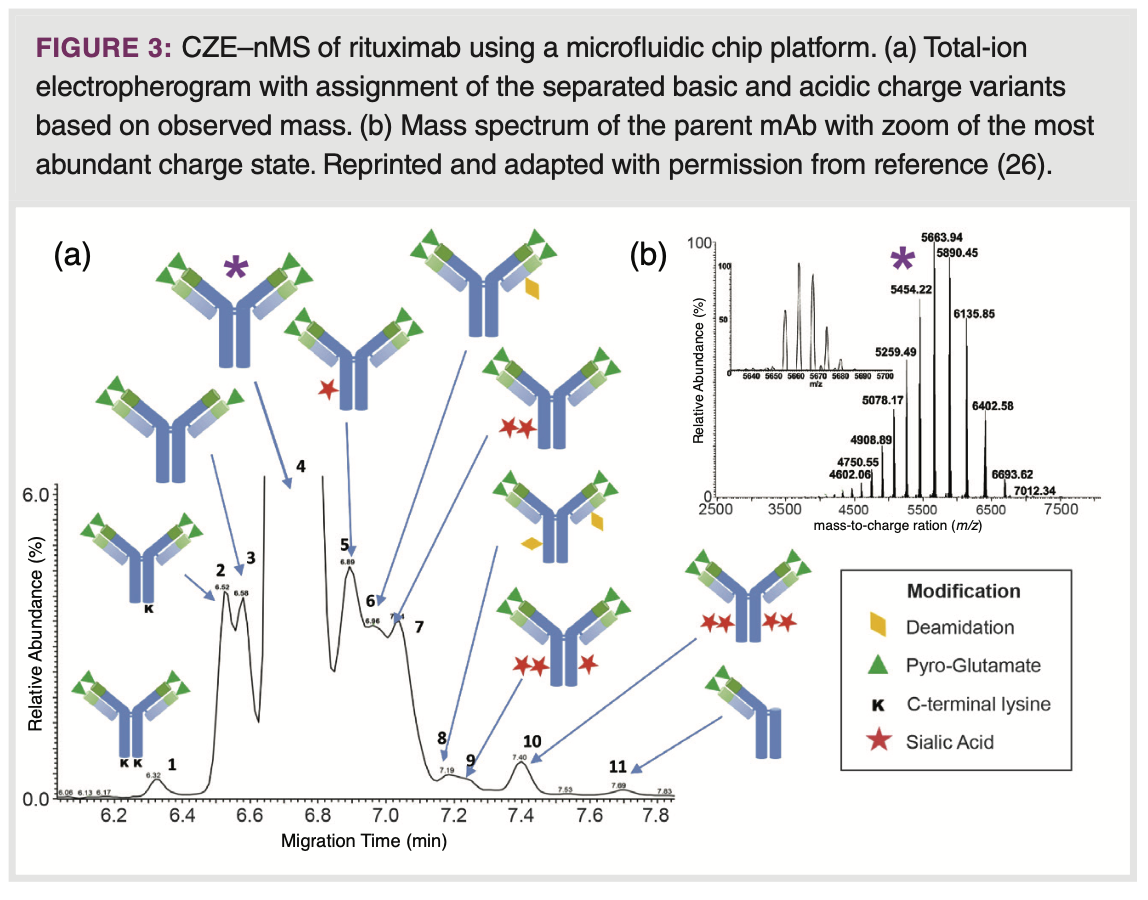
CZE–nMS: In CZE, proteins are separated in a fused-silica capillary filled with a background electrolyte (BGE) solution under the influence of an electric field. The electrophoretic mobility of a protein is proportional to the ratio of its charge and hydrodynamic radius. This property makes CZE exceptionally proficient in separating proteoforms of similar size, but different in the nature and number of charge-inducing PTMs, such as sialylation, deamidation, pyroglutamate formation and lysine clipping (23). The resolution of proteoforms is generally enhanced when the pH of the BGE is close to the isoelectric points (pIs) of the target molecules. Consequently, various CZE–UV methods for CVA use BGEs in the pH range of 5 to 7, particularly for mAb characterization. CZE–MS of intact proteins, including profiling of pharmaceutical (glyco)proteins, has been demonstrated, but frequently using acidic, denaturing conditions (24). An auspicious configuration for coupling CZE to nMS involves using a microfluidic device equipped with an integrated spray tip designed for direct electrospray ionization from the separation channel (25). Using native conditions, Carillo et al. demonstrated the effectiveness of microCZE–nMS for separating a diverse set of mAbs, including rituximab, trastuzumab, and bevacizumab, using a commercially available BGE kit (26). They achieved remarkable resolution between various charge variants, including basic proteoforms carrying non-processed C-terminal lysine, non-cyclized N-terminal glutamate, and succinimide, as well as acidic proteoforms exhibiting deamidation and sialylation (Figure 3). Wu and colleagues used a micro-CZE–nMS set up expanding the field of application to bispecific mAbs as well as therapeutic antibodies in the “N±1” format, where either one Fab arm is removed or an additional one was added to the mAb construct (27).

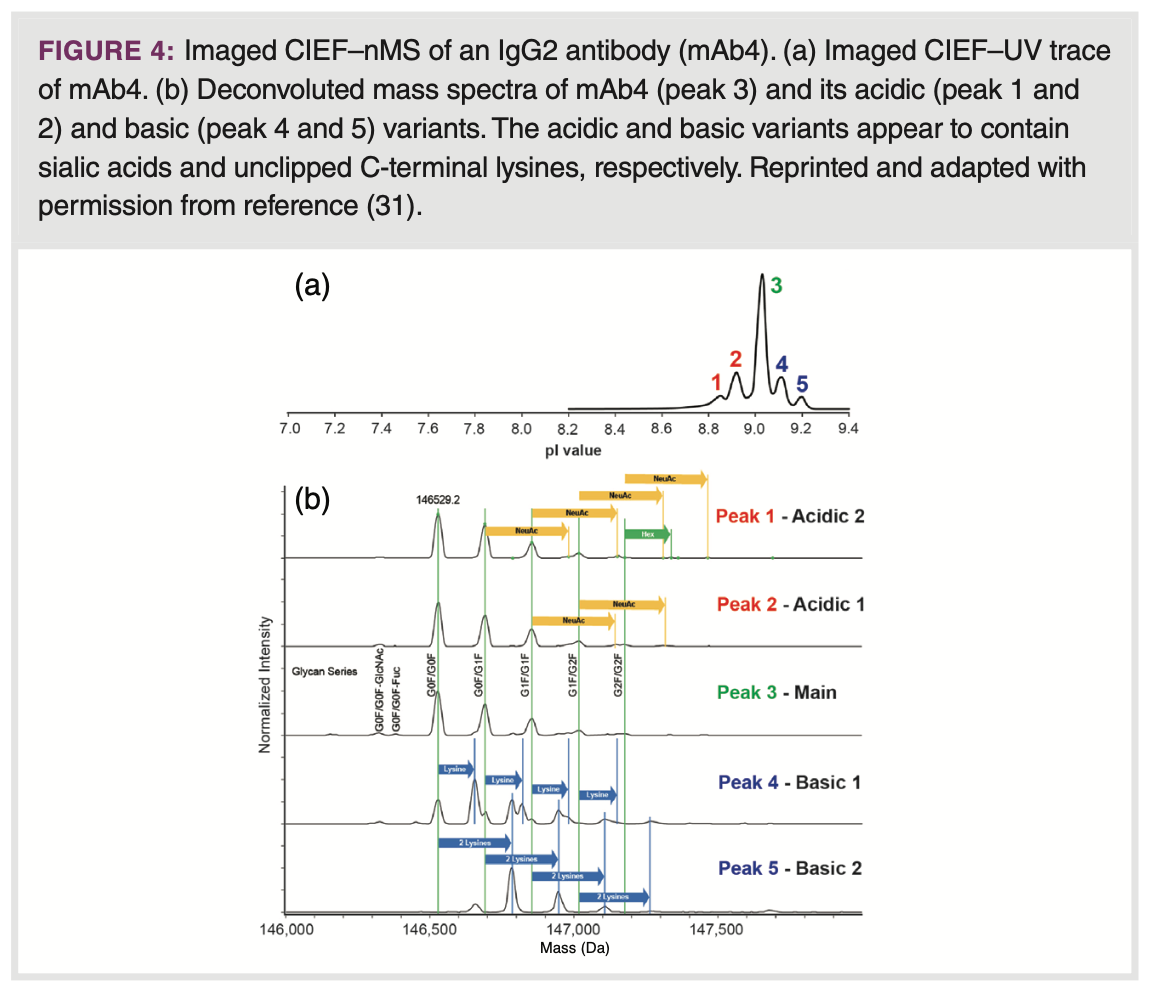
CIEF–nMS: CIEF is a technique that separates proteins according to their pI value utilizing a pH gradient. Charge-inducing PTMs noticeably impact the pI of a protein. Consequently, CIEF proves to be effective for resolving charge variants. Although CIEF, like CZE and IEX, relies on protein charge for separation, the selectivity may differ, leading to varying degrees of resolution based on the nature of the charge variants. The pH gradient is formed using a mixture of ampholytes, molecules containing both acidic and basic functional groups, defining the pH gradient range. Effective online hyphenation with MS remains a challenge, due to the ion suppression caused by ampholytes. One approach to reduce ion suppression is to directly couple CIEF to MS detection using substantially reduced amounts of ampholytes in the CIEF BGE, which will compromise protein resolution. Dai and colleagues used a nano-sheath flow-based CIEF–MS setup for the characterization of mAb charge variants, employing 1.5% v/v Pharmalyte 3-10 with 5 – 20% glycerol in the sample mixture (28). Glycerol helped to attain a stable and linear pH gradient along the migration path. Due to the application of 25 – 50% acetonitrile in the sheath liquid for CE–MS interfacing, denatured mass spectra of the mAb proteoforms were detected. Przybylski et al. characterized cytokine human interferon-gamma using CIEF–nMS by using 10 mM AmAc (pH
5) as sheath liquid (29). Typical native-like mass spectra were observed. Recently, one of the first microfluidic chip-based imaged CIEF–MS systems was launched at the 71st ASMS conference. The system allows monitoring of the mAb charge profile during the focusing step, followed by subsequent mobilization, and detection by MS. Notably, relatively high ampholyte concentrations (4% v/v) can be employed, without a distinct impact on MS sensitivity (30). Recently, the field of applications was expanded to different IgG subclasses, covering a wide range of pI values (7.3 – 9.0), and an illustrative example of the CVA of a IgG2 mAb is depicted in Figure 4 (31).

Protein Conjugates
Antibody-drug conjugates (ADCs) are produced by covalently coupling low- molecular-weight drugs to mAbs via cleavable linkers, resulting in a distribution of conjugates, typically ranging from 0 to 8 drug molecules for each mAb. The average drug-to-antibody ratio (DAR) is an essential CQA which needs to be carefully assessed. HIC has proven to be particularly valuable for DAR analysis of ADCs in which the drugs are linked to interchain cysteine residues (32). These so-called Cys-ADCs exhibit decreased structural stability compared to the parent mAb, due to the reduction of several disulfide bridges to accommodate drug attachment. The capacity of HIC to maintain native-like conditions allows to preserve the antibody structural integrity while achieving efficient conjugate separation. Online coupling of HIC to nMS is particularly intriguing in this context because it can provide accurate assignment of the respective conjugates and potential characterization of unexpected byproducts. HIC typically relies on gradients of non-volatile sulfate and phosphate salts. Substituting these with volatile alternatives is challenging, as conjugate separations are either strongly compromised or require AmAc concentrations incompatible with direct MS detection. To overcome this obstacle, Ehkirch et al. adopted a multidimensional strategy (HIC–SEC-MS) in which short-column native SEC was added as second LC dimension to attain salt exchange and MS compatibility (33). Using both untreated and artificially degraded brentuximab vedotin as model ADC, the approach allowed unambiguous profiling of the conjugate composition, including DAR. Moreover, it had the capability to characterize potential positional isomers, ADCs with non-standard DARs, and degradation products. Yan and co-workers proposed an alternative strategy to achieve HIC–nMS, involving online post-column dilution (1:6, v/v) of the 3.0-0.3 M AmAc HIC gradient with water followed by flow splitting (about 500:1), ultimately directing a flow of 1-5 μL/min to ESI-nMS (34). The utility of this approach was demonstrated by characterizing a Cys-ADC mimic under native conditions, revealing positional isomers and degraded species next to the expected conjugates. In addition, this method proved to be useful for disclosing the presence of O-linked glycan and oxidation variants in mAb samples.
Protein Affinity
Evaluating the quality and characteristics of interactions between proteoforms of a biopharmaceutical and their dedicated target is critical in drug development to understand and ensure efficacy of the product. Traditional assays for assessing affinity and binding properties typically yield a single, averaged response for a protein species (35). However, these methods do not offer the capability to differentiate between proteoforms or discern the influence of related variants on protein affinity. The combination of native protein separation with nMS can enable the investigation of structure-function and binding relationships at the proteoform level.
AC–nMS: In AC-based approaches, ligands or receptors are typically immobilized on a stationary phase (36). The retention of proteoforms is determined by their binding affinity with their respective target. AC plays a vital role in protein purification, but it can also be applied for determining protein binding characteristics. In AC, the initial mobile phase is designed to mimic physiological conditions, facilitating relevant binding interactions with the immobilized ligands or receptors, all while striving to reduce non-specific binding to a minimum. Gradually changing the pH or ionic strength of the mobile phase disrupts the interactions, eluting the proteoforms with respect to their affinity. The capabilities of AC-nMS for biopharmaceutical analysis were demonstrated through the analysis of the association of mAb proteoforms with fetal/neonatal Fc receptors (FcRn) as well as FcγRIIIa (37, 38). In both cases, the receptors were immobilized on a Sepharose stationary phase, and elution was performed applying a pH gradient. This allowed for the selective distinction of proteoforms exhibiting methionine oxidation, a common degradation, in the Fc binding domain, (37) and the influence of mAb glycosylation type on FcγRIIIa binding (38). Despite these promising results, AC–nMS faces certain limitations, including the injection of a relatively large amounts of mAb (≥50 μg), the limited pressure-resistance of AC columns, and the large amounts of receptors required to produce AC columns. These challenges might be overcome through miniaturization, potentially down to the microchip level.
ACE–nMS: ACE in combination with nMS is an auspicious approach to study non-covalent protein interactions providing proteoform resolution and identification. In ACE–MS, the ligand of interest is added to a non-denaturing BGE, and the binding receptor is injected as sample or vice versa (39). When proteoforms interact with the free ligand or receptors during CE analysis, they undergo a shift in their electrophoretic mobility. This shift is a result of the difference in charge-to-size ratio between the protein complex and the unbound protein. The extent of this shift
is governed by the affinity constant (Kd), which conceptually can be determined for each of the separated proteoform species. Unlike AC approaches, ACE does not require specific column material and only limited amount of receptor and ligand, making it more feasible for screening a variety of receptors and ligands. Additionally, ACE avoids unwanted changes in binding properties resulting from immobilization reactions (40). Conventional ACE–UV is conducted using non-volatile buffers such as sodium phosphate. In ACE–MS, these buffers are typically substituted with ammonium acetate- or bicarbonate-based BGEs at near-physiological pH. The Domínguez-Vega group utilized ACE–nMS to conduct functional studies on the binding of mAbs to FcRn (41). This approach enabled them to determine binding affinities at the individual proteoform level. Their findings revealed a reduction in mAb binding affinity in response to oxidative stress, as also indicated by a higher Kd value. Additionally, variations in glycosylation led to minor shifts in migration time, signifying that proteoforms carrying larger glycans displayed stronger binding to FcRn. The same group also investigated the binding of different mAb glycoforms to the FcγRIIa receptor by ACE-nMS (Figure 5) (42). For a mAb, exhibiting two, one, or no N-glycosylations showed that the binding affinity substantially diminished with decreasing glycosylation (Figures 5a and 5b). To confirm that the observed changes in electrophoretic mobility are indeed associated with specific receptor binding, they also examined a mutant mAb with three amino acid substitutions, resulting in silencing of the Fc region. Indeed, virtually no binding was observed between the glycoforms of the mutant mAb and FcγRIIa (Figures 5c and 5d). In addition, they unveiled, for the first time, that high mannose glycoforms exhibit diminished affinity for FcγRIIa.

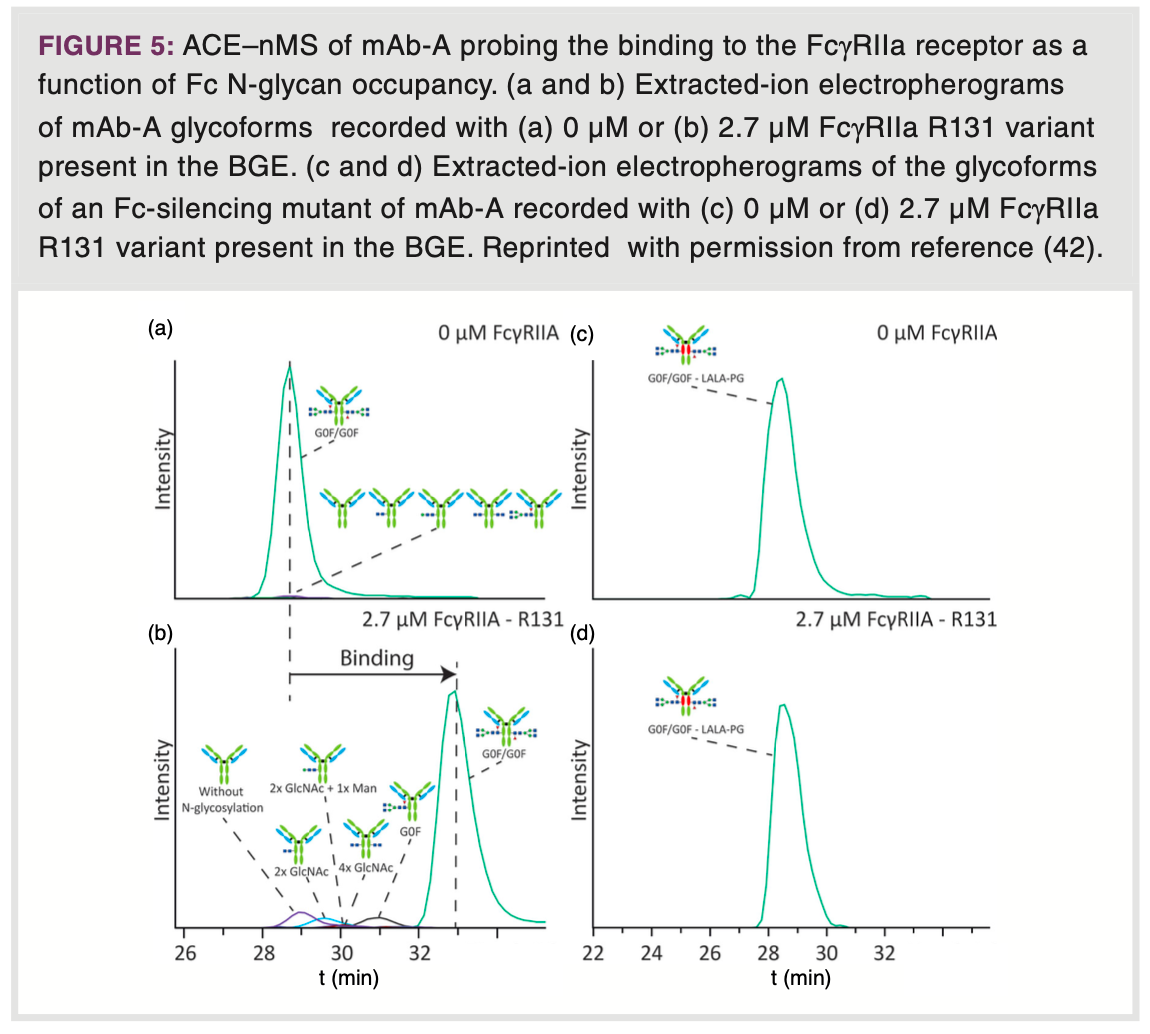
Protein Conformation
In contrast to small-molecule drugs, the conformational characteristics of protein biopharmaceuticals in solution are affected by a range of factors that go beyond their covalent chemical structures. As the conformation of a protein is closely tied to its biological function, the capacity to identify alterations is crucial in the development, purification, and formulation of a biopharmaceutical that needs to maintain consistent therapeutic properties (43).
Conformational changes of proteins do not only impact protein shape but can also have a distinct influence on net charge. In this respect, the charge-to-size separation mechanism of CZE appears eminently suitable to probe protein conformational changes under native conditions (44). In
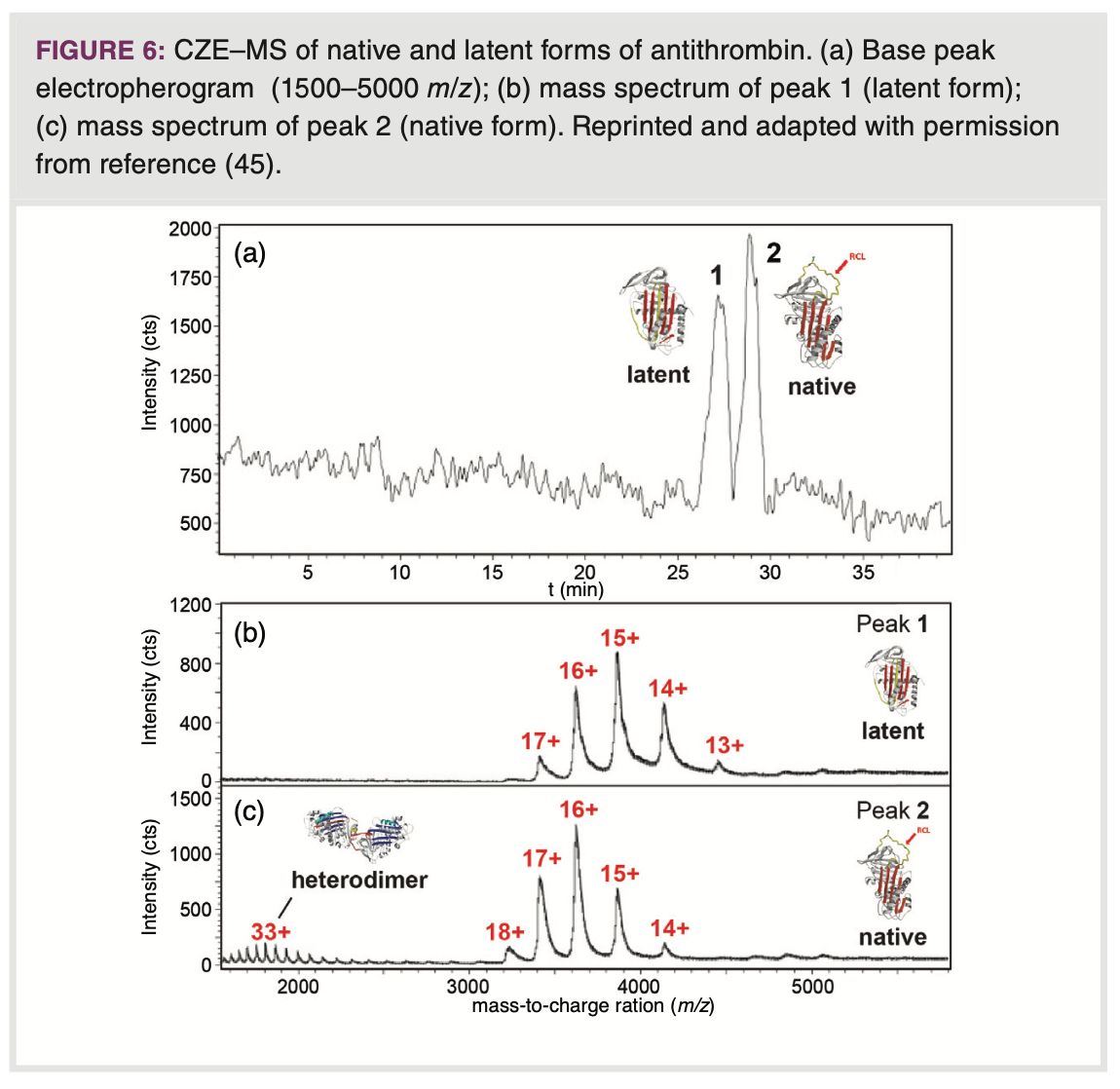
a biopharmaceutical context, CZE–MS has been used to differentiate between conformers of antithrombin (AT), a biopharmaceutical product employed for the treatment of hereditary or acquired AT deficiencies (45). Employing a BGE of 50 mM ammonium acetate (pH 7.4), CZE provided separation of the natural, active form of AT from the latent, conformationally more compact form, which exhibits no activity (Figure 6). The isomeric AT forms could be assigned as the CSD in the recorded mass spectrum of the latent form was shifted to higher m/z with respect to the active form. The CZE–MS method allowed tracking of latent form formation in AT preparations.

Conclusion
Online combinations of native protein separations with nMS have recently clearly proven to be powerful and unique means for the structural and functional characterization of biopharmaceuticals. Practical solutions have been developed to enhance the MS compatibility of native separation techniques, among which are several multidimensional approaches. 2D-LC equipment has become commercially available providing not only online desalting options, but potentially also increased resolving power by employing complementary separation selectivity. Furthermore, advancements in high-resolution MS instrumentation have made it easier to detect intact proteins in their natural state. The direct benefits of hyphenating nMS with native separation lie in the associated reduction of sample complexity, sample cleanup, and buffer exchange, ultimately resulting in protein mass spectra that yield higher-quality information. In addition, there are evident synergistic advantages to this combination. Upfront separation can facilitate deconvoluting MS signals into distinct contributing species. Conversely, the mass spectrum of a single peak may reveal the presence of multiple species, such as structural variants, aggregates, isomers, or even conformers. In the latter respect, the incorporation of ion-mobility (IM) spectrometry into native separation- nMS workflows will greatly broaden their capabilities. Over the last two decades, native IM-MS has emerged as a highly valuable tool in structural biology for the study of protein conformation and aggregation, (46) and its potential role in biopharmaceutical analysis has been recognized (47). The possibility of hyphenating native SEC and HIC to IM-MS has been demonstrated a few times, however, protein conformational information has yet to be extracted from the IM dimension (33). In coming years, there will be an expansion of the analytical toolbox with native hyphenated approaches, undoubtedly enabling more in-depth characterization of biopharmaceuticals in a shorter timeframe.
Conflicts of interest
The authors declare no conflict of interest.
References
(1) Bhatia, H.; Read, E.; Agarabi, C.; Brorson, K.; Lute, S.; Yoon, S. A Design Space Exploration for Control of Critical Quality Attributes of mAb. Int J Pharm. 2016,512 (1), 242–252. DOI: 10.1016/j.ijpharm.2016.08.046.
(2) Tamara, S.; den Boer, M. A.; Heck, A. J. R. High-Resolution Native Mass Spectrometry. Chem Rev. 2022,122 (8), 7269–7326. DOI: 10.1021/acs.chemrev.1c00212.
(3) Leney, A. C.; Heck, A. J. R. Native Mass Spectrometry: What is in the Name? J Am Soc Mass Spectr. 2017,28 (1), 5–13. DOI: 10.1007/s13361-016-1545-3.
(4) Loo, J. A. Studying Noncovalent Protein Complexes by Electrospray Ionization Mass Spectrometry. Mass Spectrom Rev. 1997,16 (1), 1–23. DOI: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L.
(5) Barth, M.; Schmidt, C. Native Mass Spectrometry-A Valuable Tool in Structural Biology. J Mass Spectrom. 2020, 55 (10). DOI: 10.1002/jms.4578.
(6) van Schaick, G.; Haselberg, R.; Somsen, G. W.; Wuhrer, M.; Domínguez-Vega, E. Studying Protein Structure and Function by Native Separation-mass Spectrometry. Nat Rev Chem. 2022,6 (3), 215–231. DOI: 10.1038/s41570-021-00353-7.
(7) Schneck, N. A.; Khatri, K.; Maust, M. D.; Jennings, M. E.; Peltier, J.; Kellie, J. F. Separation Techniques for Intact Antibody Analysis by Mass Spectrometry. J Liq Chromatogry Relat Technol. 2022, 45 (17-20), 271–283. DOI: 10.1080/10826076.2023.2199328.
(8) Schwenzer, A. K.; Kruse, L.; Jooss, K.; Neusüss, C. Capillary Electrophoresis-Mass Spectrometry for Protein Analyses Under Native Conditions. Current Progress and Perspectives. Proteomics 2023, DOI: 10.1002/pmic.202300135.
(9) Farsang, E.; Guillarme, D.; Veuthey, J. L.; Beck, A.; Lauber, M.; Schmudlach, A.; Fekete, S. Coupling Non-denaturing Chromatography to Mass Spectrometry for the Characterization of Monoclonal Antibodies and Related Products. J Pharm Biomed Anal. 2020, 185, 113207. DOI: 10.1016/j.jpba.2020.113207.
(10) Desligniére, E.; Ley, M.; Bourguet, M.; Ehkirch, A.; Botzanowski, T.; Erb, S.; Hernandez-Alba, O.; Cianferani, S. Pushing the Limits of Native MS: Online SEC-native MS for Structural Biology Applications. Int J Mass Spectrom. 2021, 461. DOI: 10.1016/j.ijms.2020.116502.
(11) Ehkirch, A.; Hernandez-Alba, O.; Colas, O.; Beck, A.; Guillarme, D.; Cianferani, S. Hyphenation of Size Exclusion Chromatography to Native Ion Mobility Mass Spectrometry for the Analytical Characterization of Therapeutic Antibodies and Related Products. J Chromatogr B Analyt Technol Biomed Life Sci. 2018,1086, 176–183. DOI: 10.1016/j.jchromb.2018.04.010.
(12) Haberger, M.; Leiss, M.; Heidenreich, A. K.; Pester, O.; Hafenmair, G.; Hook, M.; Bonnington, L.; Wegele, H.; Haindl, M.; Reusch, D.; et al. Rapid Characterization of Biotherapeutic Proteins by Size-exclusion Chromatography Coupled to Native Mass Spectrometry. Mabs-Austin 2016, 8 (2), 331–339. DOI: 10.1080/19420862.2015.1122150.
(13) VanAernum, Z. L.; Busch, F.; Jones, B. J.; Jia, M.; Chen, Z.; Boyken, S. E.; Sahasrabuddhe, A.; Baker, D.; Wysocki, V. H. Rapid Online Buffer Exchange for Screening of Proteins, Protein Complexes and Cell Lysates by Native Mass Spectrometry. Nat Protoc. 2020, 15 (3), 1132–1157. DOI: 10.1038/s41596-019-0281-0.
(14) Ventouri, I. K.; Malheiro, D. B. A.; Voeten, R. L. C.; Kok, S.; Honing, M.; Somsen, G. W.; Haselberg, R. Probing Protein Denaturation during Size-Exclusion Chromatography Using Native Mass Spectrometry. Anal Chem. 2020, 92 (6), 4292–4300. DOI: 10.1021/acs.analchem.9b04961.
(15) Murisier, A.; Andrie, M.; Fekete, S.; Lauber, M.; D’Atri, V.; Iwan, K.; Guillarme, D. Direct Coupling of Size Exclusion Chromatography and Mass Spectrometry for the Characterization of Complex Monoclonal Antibody Products. J Sep Sci. 2022, 45 (12), 1997–2007. DOI: 10.1002/jssc.202200075.
(16) Ventouri, I. K.; Veelders, S.; Passamonti, M.; Endres, P.; Roemling, R.; Schoenmakers, P. J.; Somsen, G. W.; Haselberg, R.; Gargano, A. F. G. Micro-flow Size-exclusion Chromatography for Enhanced Native Mass Spectrometry of Proteins and Protein Complexes. Anal Chim Acta. 2023, 1266, 341324. DOI: 10.1016/j.aca.2023.341324.
(17) Ventouri, I. K.; Loeber, S.; Somsen, G. W.; Schoenmakers, P. J.; Astefanei, A. Field-flow Fractionation for Molecular-Interaction Studies of Labile and Complex systems: A Critical Review. Anal Chim Acta 2022, 1193, 339396. DOI: 10.1016/j.aca.2021.339396.
(18) Ventouri, I. K.; Astefanei, A.; Kaal, E. R.; Haselberg, R.; Somsen, G. W.; Schoenmakers, P. J. Asymmetrical Flow Field-flow Fractionation to Probe the Dynamic Association Equilibria of beta-D-galactosidase. J Chromatogr A2021, 1635, 461719. DOI: 10.1016/j.chroma.2020.461719.
(19) Ventouri, I. K.; Chang, W.; Meier, F.; Drexel, R.; Somsen, G. W.; Schoenmakers, P. J.; de Spiegeleer, B.; Haselberg, R.; Astefanei, A. Characterizing Non-covalent Protein Complexes Using Asymmetrical Flow Field-Flow Fractionation On-Line Coupled to Native Mass Spectrometry. Anal Chem. 2023, 95 (19), 7487–7494. DOI: 10.1021/acs.analchem.2c05049.
(20) Auclair, J. R.; Rathore, A.; Krull, I. Charge-Variant Profiling of Biopharmaceuticals. LC GC NorthAmerica. 2018, 36 (1), 26–36.
(21) Murisier, A.; Duivelshof, B. L.; Fekete, S.; Bourquin, J.; Schmudlach, A.; Lauber, M. A.; Nguyen, J. M.; Beck, A.; Guillarme, D.; D’Atri, V. Towards a Simple On-line Coupling of Ion Exchange Chromatography and Native mass spectrometry for the Detailed Characterization of Monoclonal Antibodies. J Chromatogr A 2021, 1655, 462499 DOI: 10.1016/j.chroma.2021.462499.
(22) Haberger, M.; Heidenreich, A. K.; Hook, M.; Fichtl, J.; Lang, R.; Cymer, F.; Adibzadeh, M.; Kuhne, F.; Wegele, H.; Reusch, D.; et al. Multiattribute Monitoring of Antibody Charge Variants by Cation-Exchange Chromatography Coupled to Native Mass Spectrometry. J Am Soc Mass Spectr. 2021, 32 (8), 2062–2071. DOI: 10.1021/jasms.0c00446.
(23) Kumar, R.; Guttman, A.; Rathore, A. S. Applications of Capillary Electrophoresis for Biopharmaceutical Product Characterization. Electrophoresis 2022, 43(1-2), 143–166. DOI: 10.1002/elps.202100182.
(24) Stutz, H. Advances and Applications of Capillary Electromigration Methods in the Analysis of Therapeutic and Diagnostic Recombinant Proteins - A Review. J Pharm Biomed Anal. 222, 115089 (2023). DOI: 10.1016/j.jpba.2022.115089.
(25) Redman, E. A.; Batz, N. G.; Mellors, J. S.; Ramsey, J. M. Integrated Microfluidic Capillary Electrophoresis-Electrospray Ionization Devices with Online MS Detection for the Separation and Characterization of Intact Monoclonal Antibody Variants. Anal Chem. 2015, 87 (4), 2264–2272. DOI: 10.1021/ac503964j.
(26) Carillo, S.; Jakes, C.; Bones, J. In-depth Analysis of Monoclonal Antibodies using Microfluidic Capillary Electrophoresis and Native Mass Spectrometry. J Pharmaceut Biomed Anal. 2020, 185,DOI: 10.1016/j.jpba.2020.113218.
(27) Wu, Z. J.; Wang, H. X.; Wu, J. K.; Huang, Y.; Zhao, X. Q.; Nguyen, J. B.; Rosconi, M. P.; Pyles, E. A.; Qiu, H. B.; Li, N. High-sensitivity and High-resolution Therapeutic Antibody Charge Variant and Impurity Characterization by Microfluidic Native Capillary Electrophoresis-Mass Spectrometry. J Pharmaceut Biomed Anal.2023, 223. DOI: 10.1016/j.jpba.2022.115147.
(28) Dai, J.; Lamp, J.; Xia, Q. W.; Zhang, Y. R. Capillary Isoelectric Focusing-Mass Spectrometry Method for the Separation and Online Characterization of Intact Monoclonal Antibody Charge Variants. Anal Chem. 2018, 90 (3), 2246–2254 DOI: 10.1021/acs.analchem.7b04608.
(29) Przybylski, C.; Mokaddem, M.; Prull-Janssen, M.; Saesen, E.; Lortat-Jacob, H.; Gonnet, F.; Varenne, A.; Daniel, R. On-line Capillary Isoelectric Focusing Hyphenated to Native Electrospray Ionization Mass Spectrometry for the Characterization of Interferon-gamma and Variants. Analyst 2015, 140 (2), 543–550. DOI: 10.1039/c4an01305k.
(30) Mack, S.; Arnold, D.; Bogdan, G.; Bousse, L.; Danan, L.; Dolnik, V.; Ducusin, M.; Gwerder, E.; Herring, C.; Jensen, M.; et al. A Novel Microchip-based imaged CIEF-MS system for Comprehensive Characterization and Identification of Biopharmaceutical Charge Variants. Electrophoresis 2019, 40 (23-24), 3084–3091. DOI: 10.1002/elps.201900325.
(31) He, X. P.; ElNaggar, M.; Ostrowski, M. A.; Guttman, A.; Gentalen, E.; Sperry, J. Evaluation of an icIEF-MS System for Comparable Charge Variant Analysis of Biotherapeutics with Rapid Peak Identification by Mass Spectrometry. Electrophoresis 2022, 43 (11), 1215–1222. DOI: 10.1002/elps.202100295.
(32) Bobaly, B.; Fleury-Souverain, S.; Beck, A.; Veuthey, J. L.; Guillarme, D.; Fekete, S. Current Possibilities of Liquid Chromatography for the Characterization of Antibody-drug Conjugates. J Pharm Biomed Anal. 2018, 147, 493–505. DOI: 10.1016/j.jpba.2017.06.022.
(33) Ehkirch, A.; D’Atri, V.; Rouviere, F.; Hernandez-Alba, O.; Goyon, A.; Colas, O.; Sarrut, M.; Beck, A.; Guillarme, D.; Heinisch, S.; et al. An Online Four-Dimensional HICxSEC-IMxMS Methodology for Proof-of-Concept Characterization of Antibody Drug Conjugates. Anal Chem. 2018, 90 (3), 1578–1586. DOI: 10.1021/acs.analchem.7b02110.
(34) Yan, Y.; Xing, T.; Wang, S.; Daly, T. J.; Li, N. Online Coupling of Analytical Hydrophobic Interaction Chromatography with Native Mass Spectrometry for the Characterization of Monoclonal Antibodies and Related Products. J Pharm Biomed Anal. 2020, 186, 113313. DOI: 10.1016/j.jpba.2020.113313.
(35) Suh, K.; Kyei, I.; Hage, D. S. Approaches for the Detection and Analysis of Antidrug Antibodies to Biopharmaceuticals: A Review. J Sep Sci. 2022, 45 (12), 2077–2092. DOI: 10.1002/jssc.202200112.
(36) Rodriguez, E. L.; Poddar, S.; Iftekhar, S.; Suh, K.; Woolfork, A. G.; Ovbude, S.; Pekarek, A.; Walters, M.; Lott, S.; Hage, D. S. Affinity Chromatography: A Review of Trends and Developments Over the Past 50 years. J Chromatogr B 2020, 1157,DOI: 10.1016/j.jchromb.2020.122332.
(37) Gahoual, R.; Heidenreich, A. K.; Somsen, G. W.; Bulau, P.; Reusch, D.; Wuhrer, M.; Haberger, M. Detailed Characterization of Monoclonal Antibody Receptor Interaction Using Affinity Liquid Chromatography Hyphenated to Native Mass Spectrometry. Anal Chem. 2017, 89 (10), 5404–5412. DOI: 10.1021/acs.analchem.7b00211.
(38) Lippold, S.; Nicolardi, S.; Domínguez-Vega, E.; Heidenreich, A. K.; Vidarsson, G.; Reusch, D.; Haberger, M.; Wuhrer, M.; Falck, D. Glycoform-Resolved FcγRIIIa Affinity Chromatography-Mass Spectrometry. Mabs-Austin 2019, 11 (7), 1191–1196 DOI: 10.1080/19420862.2019.1636602.
(39) Chen, Z.; Weber, S. G. Determination of Binding Constants by Affinity Capillary Electrophoresis, Electrospray Ionization Mass Spectrometry and Phase-distribution Methods. Trac-Trend Anal Chem. 2008, 27 (9), 738–748. DOI: 10.1016/j.trac.2008.06.008.
(40) Dubsky, P.; Dvorák, M.; Ansorge, M. Affinity capillary Electrophoresis: the Theory of Electromigration. Anal Bioanal Chem. 2016, 408 (30), 8623–8641. DOI: 10.1007/s00216-016-9799-y.
(41) Gstöttner, C.; Hook, M.; Christopeit, T.; Knaupp, A.; Schlothauer, T.; Reusch, D.; Haberger, M.; Wuhrer, M.; Domínguez-Vega, E. Affinity Capillary Electrophoresis-Mass Spectrometry as a Tool to Unravel Proteoform-Specific Antibody-Receptor Interactions. Anal Chem.2021, 93(45), 15133–15141. DOI: 10.1021/acs.analchem.1c03560.
(42) Gstöttner, C.; Knaupp, A.; Vidarsson, G.; Reusch, D.; Schlothauer, T.; Wuhrer, M.; Domínguez-Vega, E. Affinity Capillary Electrophoresis - Mass Spectrometry Permits Direct Binding Assessment of IgG and FcγRIIa in a Glycoform-resolved Manner. Front Immunol. 2022,13. DOI: 10.3389/fimmu.2022.980291.
(43) Bobst, C. E.; Abzalimov, R. R.; Houde, D.; Kloczewiak, M.; Mhatre, R.; Berkowitz, S. A.; Kaltashov, I. A. Detection and Characterization of Altered Conformations of Protein Pharmaceuticals using Complementary Mass Spectrometry-based Approaches. Anal Chem. 2008,80 (19), 7473–7481. DOI: 10.1021/ac801214x.
(44) Zhang, W. J.; Xiang, Y.; Xu, W. Probing Protein Higher-order Structures by Native Capillary Electrophoresis-Mass Spectrometry. Trac-Trend Anal Chem, 2022,157.DOI: 10.1016/j.trac.2022.116739.
(45) Marie, A. L.; Dominguez-Vega, E.; Saller, F.; Plantier, J. L.; Urbain, R.; Borgel, D.; Tran, N. T.; Somsen, G. W.; Taverna, M. Characterization of Conformers and Dimers of Antithrombin by Capillary Electrophoresis-quadrupole-time-of-flight Mass Spectrometry. Anal Chim Acta, 2016, 947, 58–65. DOI: 10.1016/j.aca.2016.10.016.
(46) Christofi, E.; Barran, P. Ion Mobility Mass Spectrometry (IM-MS) for Structural Biology: Insights Gained by Measuring Mass, Charge, and Collision Cross Section. Chem Rev, 2023,123 (6), 2902–2949. DOI: 10.1021/acs.chemrev.2c00600.
(47) Skeene, K.; Khatri, K.; Soloviev, Z.; Lapthorn, C. Current status and Future Prospects for Ion-mobility Mass spectrometry in the Biopharmaceutical Industry. Biochim Biophys Acta Proteins Proteom, 2021, 1869 (12), 140697. DOI: 10.1016/j.bbapap.2021.140697.
Kevin Jooß is assistant professor at the Division of Bioanalytical Chemistry of Vrije Universiteit Amsterdam, The Netherlands. His research focus is on intact and top-down protein analysis using native mass spectrometry and CE–MS technology with applications in molecular biology and biopharmaceutical analysis.
Govert W. Somsen is full professor in Biomolecular Analysis at Vrije Universiteit Amsterdam, The Netherlands. His primary research interest lies in intact and native biopolymer analysis developing and applying high-resolution separation techniques hyphenated with (ion-mobility) mass spectrometry. He is editor of Journal of Chromatography B.





 Xolair technical development team leader from Phase 2 through licensing. Reed is a CASSS Distinguished Fellow and has been an author of 49 publications, including 14 as corresponding author. He was a leader in the development of Roche-Genentech’s strategies for post-approval comparability exercises, clinical and commercial specifications, the identification of critical quality attributes, linking structure to function, the use of a risk-based Quality by Design approach for control strategy development, unexpected protein modification identification, and curiosity-based thinking.")






. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.


