Ion-Mobility Mass Spectrometry in Metabolomics and Lipidomics
LCGC Europe
Ion-mobility spectrometry (IMS) is an analytical technique that provides for the separation of ions in the gas phase. The separation, occuring on a timescale of milliseconds, is based on the differing mobility of ions according to their charge, shape, and size. These characteristics make IMS suitable for coupling with mass spectrometry (MS), to serve in current MS-based workflows for metabolomics and lipidomics. IMS–MS improves peak capacity and signal-to-noise ratios, and it provides more confidence during compound identification or confirmation than conventional analyses.
Ion-mobility spectrometry (IMS) is an analytical technique that provides for the separation of ions in the gas phase. The separation, occuring on a timescale of milliseconds, is based on the differing mobility of ions according to their charge, shape, and size. These characteristics make IMS suitable for coupling with mass spectrometry (MS), to serve in current MS-based workflows for metabolomics and lipidomics. IMS–MS improves peak capacity and signal-to-noise ratios, and it provides more confidence during compound identification or confirmation than conventional analyses. Combining collision-induced dissociation with ion-mobility separation improves the specificity of MS–MS-based approaches. Here, we discuss these techniques as well other recent advances in this area.
Ion-mobility spectrometry (IMS) provides for the separation of gasâphase ions according to their mass-to-charge ratio (m/z) and shape (1,2). The separation occurs on a timescale of milliseconds, making it suitable for coupling with mass spectrometry (MS), in which detection with time-of-flight (TOF) instruments usually occurs on a microsecond scale.
Three commercial IMS–MS technologies are currently available (1–3): drift-time ion-mobility spectrometry (DT–IMS), travelling-wave ion-mobility spectrometry (TW–IMS), and differential-mobility spectrometry (DMS), the latter of which is also called field–asymmetric ion-mobility spectrometry (FAIMS). In DT–IMS, ions migrate through a buffer gas in the presence of an axial, linear, electricâfield gradient. In TW–IMS, a sequence of applied voltages generates a so-called travelling wave that propels the ions through the buffer gas. Thus, both DT–IMS and TW–IMS allow all ions to pass through the mobility cell. Alternatively, DMS operates by varying compensation voltage, to filter selected ions in a space-dispersive fashion (1,2).
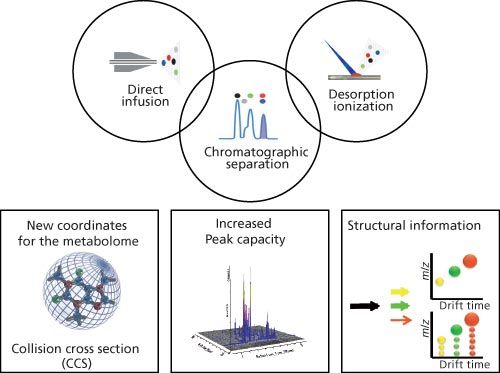
In recent years, exploiting both hardware and software advances, IMS–MS has been increasingly used in traditional metabolomic and lipidomic approaches including liquid chromatography (LC)–MS and direct-MS analyses, such as shotgun lipidomics and MS imaging (Figure 1) (4–8).


Figure 1: Diagram showing the three major benefits of ion mobility to traditional MS-based metabolomics and lipidomics workflows: it provides the CCS of a given metabolite, increases the peak capacity, and, when coupled with fragmentation, provides a new level of structural information.
Ion-Mobility-Derived Collision Cross Sections
The drift time, which is the time required for ions to cross the ionâmobility separation cell, depends on the collision frequency between an ion and the buffer gas. Thus, the drift time is intrinsically linked to the shape, size, and charge of a given ion as well as to the nature of the buffer gas. From the drift time, you can calculate the rotationally averaged collision cross section (CCS), which represents the effective area for the interaction between an individual ion and the neutral gas through which it travels. CCS, an important physicochemical property of lipids and metabolites, is related to chemical structure and three-dimensional (3D) conformation.
In traditional DT–IMS devices, which provide a uniform electrostatic field, you can derive CCS directly from the drift time (9). In TW–IMS, the applied electric field waveform function is more complex than the drift tube used in the traditional devices. CCS values can be automatically calculated from drift-time data by referring to compounds of known CCS values, such as polyalanine (10). This calibration corrects for the variation in drift time among IMS–MS instruments in independent laboratories, where ion-mobility parameter settings vary, resulting in highly reproducible CCS measurements (4,10).
IMS–MS in Direct-Infusion Experiments
The direct infusion of metabolite and lipid extracts into the ionization source of a mass spectrometer enables high-throughput analysis of biological samples. However, the approach is associated with the inability to distinguish isobaric species (11). Using IMS–MS in combination with directâinfusion experiments could - at least partially - overcome this drawback. IMS–MS can separate metabolites and lipids in two dimensions according to their mobility times and masses. In doing so, it disassociates them from other classes of biomolecules such as peptides, carbohydrates, and oligosaccharides (12). What’s more, including ion mobility in a direct-infusion experiment may provide as much as a fivefold increase in peak capacity (5).
On-Line Chromatographic Separation–IMS–MS
When coupled with MS, chromatographic separations like gas chromatography (GC), ultrahigh-pressure LC (UHPLC), and supercritical fluid chromatography (SFC) might offer a valuable solution to resolving complex mixtures of analytes. But biological samples contain several thousand metabolites and lipids, which creates a challenge for chromatography to completely meet. The additional degree of ionâmobility separation gained by coupling LC with IMS increases peak capacity by distinguishing metabolites from matrix interferences, and thus helps to generate cleaner mass-spectral data and even provides the ability to resolve isobars and structural isomers (Figure 2) (13). Despite a tendency towards shifting retention-time values (because of samples loading and matrix effect), IMS information confers a considerable benefit in the form of enhanced reproducibility of ionâmobility-derived CCS measurements, which provide an additional coordinate for metabolite and lipid identification. IMS–MS thus improves confidence in metabolomics studies by reducing the number of potential false negative and false positive identifications (Figure 3) (4,5).

Figure 2: Separation of isomeric and isobaric species. Adding ion mobility to a typical workflow increases the peak capacity of the analytical workflow by separating somers and isobars of lutein extracted from algae in gas phase that would otherwise be coeluted (13).
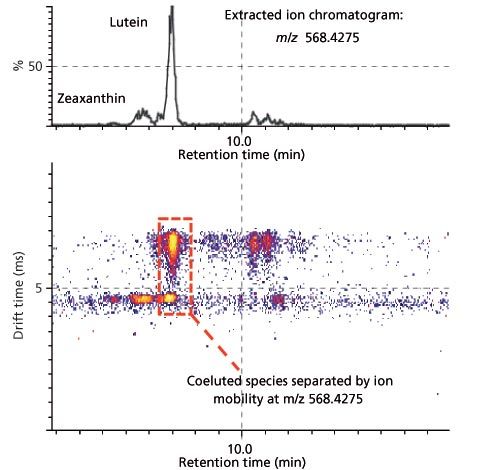

Figure 3: A typical UHPLC–TW–IMS–MS workflow can be divided into three steps: analytical measurement, data processing, and metabolite identification. The analysis provides three specific coordinates (CCS, mass, and Kd) that are characteristic of a given molecule and can be used for metabolite identification.

In qualitative and quantitative applications, novel informatics technologies can now process on-line chromatographically separated IMS–MS data. A typical LC–, SFC–, or GC–IMS–MS metabolomic or lipidomic experiment automatically generates a list of metabolite annotations. The list includes, as molecular identifiers, CCS values as well as retentionâtime and mass (m/z) values (Figure 3) (4,5). To identify analytes, you can search experimentally derived CCS values against databases (4) created using CCS values obtained by running synthetic standards or by computational calculation. The difference between reference CCS (contained in the database) and experimental CCS values (DCCS) can contribute to the identification score. You can search for metabolites by selecting a DCCS that is less than the DCCS of a given threshold (4,5). In this way, you can filter and score identifications when querying the database containing the CCS information, reducing the number of false positives and false negatives (4,5).
IMS–MS for Data-Independent Acquisition
To identify metabolites, you must often interpret fragmentation spectra obtained using various MS–MS modes of acquisition. A data-independent mode of acquisition that alternates between low and high collision energies has proved effective in lipidomics and metabolomics analyses when combined with on-line chromatographic separation (4–6). In one application, the data-independent acquisition mode provides accurateâmass measurement of precursor ions from the low-collisionâenergy data, and it provides product ions from the elevatedâcollisionâenergy data (4,5). Subsequently, the product ions are matched according to retention-time correlation data with the appropriate precursor ions (4,5). Yet the results are not always ideal. Applying LC–MS in data-independent acquisition mode to lipid and metabolite extracts from biological samples often produces spectra that reflect mixed product ions derived from many coeluting precursor ions. The mixed nature of these product ions complicates spectral interpretation and, therefore, the overall identification process. IMS–MS, however, enhances the data-independent acquisition process. Incorporating ion mobility as part of the data-independent acquisition process gives rise to a new acquisition mode, wherein coeluted precursor ions are separated by ion-mobility before fragmenting, resulting in MS–MS spectra in which the product ions are relatively homogenous (4–7). Drift times offer additional deconvolution information, further associating product ions with their coeluting precursor ions. The clean MS–MS spectra that result facilitate metabolite and lipid identification by increasing specificity and reducing falseâpositive assignments (4–7).
Coupling Pre- and Post-Mobility Fragmentation: Time-Aligned Parallel Fragmentation
Owing to their complexity and structural variety, many metabolite and lipid structures tend to resist characterization by MS–MS. IMS–MS, however, offers a pseudo-MS3 mode of acquisition: timeâaligned parallel (TAP) fragmentation. By means of the mass spectrometer’s quadrupole mass filter, TAP makes it possible to select a particular ion of interest. That ion is then fragmented in the instrument’s first collision cell. The resultant fragmentation ions, separated according to ion mobility, are then further fragmented in a second collision cell, located after the travelling-wave ion mobility cell. This mode of acquisition has been used to characterize, in a single analytical step, the structure of complex metabolites and lipids (6).
IMS–MS Imaging
MS imaging confers the ability to topographically map the metabolites and lipid content across the surface of biological samples such as cell cultures and tissue sections. The technique involves irradiating a section of frozen tissue using a focused excitatory beam such as a laser, ions, or droplets of charged solvent and then scanning a selected area of the tissue along all axes (7,17,18). On impact, the sample surface releases a vapour of ionized molecules that are then induced into the detector, for accurate-mass measurement. Adding the dimension of ion mobility to this experiment facilitates detection of smallâmolecule metabolites and lipids (5,7). It helps by separating many isobaric species, as well as matrix and peptides, and by improving the overall sensitivity, signal-to-noise ratio, and specificity of MS imaging. Besides ascertaining accurate mass, you can search the experimental CCS of each detected ion against CCS databases, to support identification (4,5,7,19). Various desorption ionization techniques have been combined with IMS–MS, for imaging of metabolites and lipids. They include matrixâassisted laser desorption-ionization (MALDI) (7), desorption electrospray ionization (DESI), direct analysis in real time (DART), and laser-ablation electrospray ionization (LAESI) (20).
Conclusions and Future Directions
Hardware and software enhancements allow integration of ion-mobility mass spectrometry with the traditional metabolomics approaches of direct infusion, on-line chromatographic separation, and desorption ionization. Increasingly, therefore, IMS–MS is being adopted for use in metabolomics and lipidomics applications. Combining ion mobility with those traditional analytical approaches offers three main advantages for metabolomics. First, IMS allows for measuring CCS. A physicochemical identifier relating to the shape of lipids and metabolites while in the gas phase, CCS adds an additional physical-chemical coordinate for lipid and metabolite identification, the others being retention time and mass. Second, an IMS post-ionization separation increases peak capacity and the signalâto-noise ratio of complex mixtures of metabolites and lipids present in biological samples, improving molecular fingerprinting and spatial localization. Finally, IMS lends itself to coupling with various MS–MS fragmentation acquisition modes, adding a new set of tools for structural characterization that improves the specificity of lipid and metabolite identification.
We expect that incorporating IMS–MS technology into traditional analytical workflows will significantly benefit analysts facing the challenges of a metabolomics and lipidomics experiment.
References
- A.B. Kanu, P. Dwivedi, M. Tam, L. Matz, and H.H. Hill Jr., J. Mass Spectrom.43(1), 1–22 (2008). doi:10.1002/jms.1383
- J.C. May and J.A. McLean, Anal. Chem. 87(3), 1422–1436 (2015). doi:10.1021/ac504720m
- K. Giles, J.P. Williams, and I. Campuzano, Rapid Commun. Mass Spectrom.25(11), 1559–1566 (2011). doi:10.1002/rcm.5013
- G. Paglia, J.P. Williams, L. Menikarachchi, J.W. Thompson, R. Tyldesley-Worster, S. Halldorsson, O. Rolfsson, A. Moseley, D. Grant, J. Langridge, B.O. Palsson, and G. Astarita, Anal. Chem.86(8), 3985–3993 (2014). doi:10.1021/ac500405x
- G. Paglia, P. Angel, J.P. Williams, K. Richardson, H.J. Olivos, J.W. Thompson, L. Menikarachchi, S. Lai, C. Walsh, A. Moseley, R.S. Plumb, D.F. Grant, B.O. Palsson, J. Langridge, S. Geromanos, and G. Astarita, Anal. Chem. 87(2), 1137–1144 (2015). doi:10.1021/ac503715v
- J. Castro-Perez, T.P. Roddy, N.M. Nibbering, V. Shah, D.G. McLaren, S. Previs, A.B. Attygalle, K. Herath, Z. Chen, S.P. Wang, L. Mitnaul, B.K. Hubbard, R.J. Vreeken, D.G. Johns, and T. Hankemeier, J. Am. Soc. Mass Spectrom.22(9), 1552–1567 (2011). doi:10.1007/s13361-011-0172-2
- P.J. Hart, S. Francese, E. Claude, M.N. Woodroofe, and M.R. Clench, Anal. Bioanal. Chem. 401(1), 115–125 (2011). doi:10.1007/s00216-011-5090-4
- P. Dwivedi, A.J. Schultz, and H.H. Hill, Int. J. Mass Spectrom.298(1–3), 78–90 (2010). doi:10.1016/j.ijms.2010.02.007
- E.A. Mason and E.W. McDaniel, Transport Properties of Ions in Gases (John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 1998) doi:10.1002/3527602852.fmatter.
- M.F. Bush, I.D. Campuzano, and C.V. Robinson, Anal. Chem. 84(16), 7124–7130 (2012). doi:10.1021/ac3014498
- M. Kliman, J.C. May, and J.A. McLean, Biochim. Biophys. Acta1811(11), 935–945 (2011). doi:10.1016/j.bbalip.2011.05.016
- S.N. Jackson, M. Ugarov, J.D. Post, T. Egan, D. Langlais, J.A. Schultz, and A.S. Woods, J. Am. Soc. Mass Spectrom. 19(11), 1655–1662 (2008). doi:10.1016/j.jasms.2008.07.005
- V. Shah, J.M. Castro-Perez, D.G. McLaren, K.B. Herath, S.F. Previs, and T.P. Roddy, Rapid Commun. Mass Spectrom.27(19), 2195–2200 (2013). doi:10.1002/rcm.6675
- W.P. Chong, L.T. Goh, S.G. Reddy, F.N. Yusufi, D.Y. Lee, N.S. Wong, C.K. Heng, M.G. Yap, and Y.S. Ho, Rapid Commun. Mass Spectrom.23(23), 3763–3771 (2009). doi:10.1002/rcm.4328
- G.B. Gonzales, K. Raes, S. Coelus, K. Struijs, G. Smagghe, and J. Van Camp, J. Chromatogr. A1323, 39–48 (2014). doi:10.1016/j.chroma.2013.10.077
- S. Garmón-Lobato, B. Abad-García, M.B. Sánchez-Ilárduya, M. Romera-Fernández, L.A. Berrueta, B. Gallo, and F. Vicente, Anal. Chim. Acta 10(771), 56–64 (2013). doi: 10.1016/j.aca.2013.01.065
- P.J. Trim, S.J. Atkinson, A.P. Princivalle, P.S. Marshall, A. West, and M.R. Clench, Rapid Commun. Mass Spectrom. 22(10), 1503–1509 (2008). doi:10.1002/rcm.3498
- M.C. Djidja, E. Claude, M.F. Snel, S. Francese, P. Scriven, V. Carolan, and M.R. Clench, Anal. Bioanal. Chem.397(2), 587–601 (2010). doi:10.1007/s00216-010-3554-6
- T. Pacini, W. Fu, S. Gudmundsson, A.E. Chiaravalle, S. Brynjolfson, B.O. Palsson, G. Astarita, and G. Paglia, Anal. Chem.87(5), 2593–2599 (2015). doi:10.1021/ac504707n
- H. Li, B. K. Smith, L. Márk, P. Nemes, J. Nazarian, and A. Vertes, Int. J. Mass Spectrom.377, 681–689 (2015). doi.org/10.1016/j.ijms.2014.06.025
Giuseppe Paglia is a researcher at the Istituto Zooprofilattico di Puglia e Basilicata, where he is head of the metabolomics facility. Paglia’s research interests include MS-based lipidomics and metabolomics, as applied to food safety and biomedical application.
Giuseppe Astarita is a principal scientist at Waters Corporation in Milford, Massachusetts, USA, and serves as adjunct professor at Georgetown University in Washington, D.C. Giuseppe’s research interests include lipid mass spectrometry and metabolomics, as applied to translational medicine and biomarker discovery. He is applying the latest technologies in the study of health and disease as well as food and nutrition. Direct correspondence to: giuseppe_astarita@waters.com
“MS - The Practical Art” Editor Kate Yu joined Waters in Milford, Massachusetts, USA, in 1998. She has a wealth of experience in applying LC–MS technologies to various application fields such as metabolite identification, metabolomics, quantitative bioanalysis, natural products, and environmental applications. Direct correspondence about this column should be sent to the editorâin-chief, Alasdair Matheson, at amatheson@advanstar.com











Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

Determining the Serum Proteomic Profile in Migraine Patients with LC–MS
April 17th 2025Researchers used liquid chromatography–mass spectrometry (LC–MS) in their proteomic analysis to compare the serum proteome of migraine patients with healthy controls and to identify differentially expressed proteins as potential migraine biomarkers.

