Improved Sensitivity and Specificity for trans-Resveratrol in Red Wine Analysis with HPLC–UV and LC–MS
LCGC Europe
Adequate detection of trans-resveratrol in wine is complicated by two factors: relatively low levels and interferences from matrix components. Here, we present two useful approaches to overcoming these issues depending on the instrumentation available. For high performance liquid chromatography (HPLC) analyses with ultraviolet (UV) detection, matrix peaks can be removed by microextraction using a packed sorbent while simultaneously concentrating the trans-resveratrol peak by a factor of two. For liquid chromatography–mass spectrometry (LC–MS), the extracted ion chromatogram for the [M + H]+ analyte ion can be used to obtain specificity without prior extraction procedures.


Photo Credit: chris van dolleweerd/Getty Images

Joshua E. Young1, Michael V. Lim2, Joshua Topete2, Hao Hang2, Marion Gahol2, Joseph J. Pesek2, and Maria T. Matyska2, 1MicroSolv Technology Corporation, Eatontown, New Jersey, USA, 2San José State University, San José, California, USA.
Adequate detection of trans-resveratrol in wine is complicated by two factors: relatively low levels and interferences from matrix components. Here, we present two useful approaches to overcoming these issues depending on the instrumentation available. For high performance liquid chromatography (HPLC) analyses with ultraviolet (UV) detection, matrix peaks can be removed by microextraction using a packed sorbent while simultaneously concentrating the trans-resveratrol peak by a factor of two. For liquid chromatography–mass spectrometry (LC–MS), the extracted ion chromatogram for the [M + H]+ analyte ion can be used to obtain specificity without prior extraction procedures.
Resveratrol is a naturally occurring stilbenoid compound found in certain plant components, such as grape skins, peanuts, and berries. Although the practical extent of its therapeutic effects is debated, resveratrol has been variously reported to exhibit cardio-protective (1), anticancer (2), and neuroprotective (3) properties. A common point of contention with findings regarding the purported health benefits of resveratrol is that many such studies are performed in vitro or with animal subjects (4). Hence, the conclusions may not necessarily translate to in vivo human clinical trials, which are still relatively scarce at present (5). For example, the low bioavailability of resveratrol may not allow for the relatively high concentrations used to achieve significant efficacy in some in vitro studies (6).
Nevertheless, the amount of resveratrol in food and beverage products is of ongoing interest to producers and consumers. Red wine, a common food source of resveratrol, is therefore pertinent to analytical study in furtherance to these investigations. The content of resveratrol can vary widely among red wine samples depending on factors such as region of cultivation and grape variety, but reported values have ranged up to 14.3 mg/L (7). With ultraviolet (UV) detection in high performance liquid chromatography (HPLC) analyses, the lower levels found in some samples may be somewhat difficult to adequately quantitate. Furthermore, peaks from other components in the wine extracts may interfere with resveratrol detection. Liquid chromatography coupled to mass spectrometry (LC–MS) has been shown to be a powerful tool capable of detecting trans-resveratrol down to 0.3 pmol (8) and can also differentiate based on the mass-toâcharge ratio (m/z), but many laboratories may lack this more sophisticated instrumentation. For analyses with UV-based instruments, a sample cleanup strategy is generally required before HPLC analysis.
In recent years, miniaturized solid-phase extraction (SPE) techniques have become an advantageous alternative to traditional sample cleanup or preconcentration procedures (9). Using conventional liquid–liquid extraction (LLE), relatively large amounts of solvent are typically required for a single sample, which increases cost for both solvent usage and disposal. The process is also not amenable to automation, taking up valuable analyst time. SPE greatly reduces solvent consumption compared to LLE, but can be attenuated further with more modern microextraction techniques. In addition to solvent savings, miniaturized SPE approaches require very low amounts of sample (on the order of microlitres versus millilitres for SPE), which can be beneficial if the sample is expensive or scarce.
Microextraction by packed sorbent can be thought of as a type of miniaturized SPE technique where the solid phase is a small packed bed (~1–2 mg) incorporated into the barrel of a pipettete needle. Carryover from previous samples using the same cartridge can be reduced or eliminated compared to traditional SPE because of the smaller sorbent bed and lower wash volumes involved (10). The process is also generally faster and more readily automated than SPE or LLE, since the packed sorbent bed is part of the syringe (11).
The extraction technique entails the same general principles as chromatography, with a few notable caveats. Because the process must be quantitative to be of use, extremes in retention or elution behaviour are preferable. A sample is loaded onto the cartridge under strongly retaining conditions such that the analyte of interest will be quantitatively adsorbed onto the sorbent. Strongly eluting conditions are used to then completely remove the analyte from the packed bed and into the sample vial for analysis. Since the sorbent is smaller in microextraction by packed sorbent than in traditional SPE, greater attention must be paid to ensure the analyte loading capacity of the packed bed is not exceeded.
There are two primary reasons to incorporate a microextraction by packed sorbent procedure into sample preparation. First, compounds that are not retained on the sorbent are removed from the final sample, which might otherwise show up as interfering peaks in the subsequent HPLC chromatograms. Hence, the technique can improve the specificity of the analysis. Second, if the volume of liquid used in the elution step is lower than that of the loading step, an increase in analyte concentration is obtained. This can be performed by using one large volume in the loading step or through a series of extract–discard cycles of equal volume to the elution step. If an analyte is generally found in low levels in a particular sample, this microextraction by packed sorbent preconcentration technique may be an effective strategy to adequately detect and quantitate it. The resulting concentration increase is then accounted for in the analytical calculations, allowing for the quantitation of analytes present at levels below the linear range of the calibration curve.
In the present work, microextraction by packed sorbent approaches were investigated as a means to increase the concentration of resveratrol in red wine samples while simultaneously removing undesirable compounds from the extracts. A silica hydride-based C18 material was used as the sorbent in the cartridges and compared to an analogous C18 phase based on conventional silica. To our knowledge, this is the first instance of silica hydride sorbents used for a microextraction sample preconcentration application. Previous work on the utility of these materials as analytical HPLC columns in food and beverage analyses has been demonstrated for limonin in orange juice (12) and histamine in foodstuffs (13). Silica hydride-based sorbents have also been used in traditional SPE in an application analyzing components of dark chocolate (14).
One notable study (15) reported the use of microâextraction by packed sorbent in analysis of trans-resveratrol in wine samples, but in that case a preconcentration factor of the extract was not used in the final method. Preconcentration had been investigated through the use of multiple extract–discard cycles of sample aliquots on the cartridges, and increases in peak areas were observed, but the results were not quantitative (for example, the use of five extraction cycles in the microextraction by packed sorbent preconcentration step in sample preparation yielded significantly less than a fivefold increase in peak area in the HPLC data, relative to that of the single extraction cycle). If the increase in concentration is to be accurately accounted for in the calculations, the peak area must be directly proportional to the preconcentration factor. Hence, another aspect of this study was to investigate the preconcentration aspect of the microextraction by packed sorbent technique more thoroughly.
Because of the inherent sensitivity and specificity capabilities of LC–MS, its use was investigated for analysis of trans-resveratrol in red wine as an alternative strategy to the microextraction by packed sorbent approach. This may be pertinent to researchers investigating resveratrol at very low levels or in more complex samples such as plasma extracts. Analysts can isolate the extracted ion chromatogram (EIC) corresponding to the [M + H]+ ion for resveratrol, leading to superior specificity compared to UV-based analyses. As such, extraction-based sample cleanup procedures may be unnecessary with the use of this more sophisticated instrumentation.
Materials and Methods
Materials: Resveratrol capsules and red wine (Merlot, 2012, cultivated in California, USA) were purchased from local supermarkets. Formic acid and a transâresveratrol reference standard were from Sigma–Aldrich. Acetonitrile, isopropanol, methanol, and deionized water (HPLC grades) were obtained from GFS Chemicals, Inc.
Instrumentation:
HPLC–UV: A Hewlett-Packard 1100 HPLC system consisting of an autosampler, degasser, binary pump, and variable wavelength UV detector (304 nm) was used. The system was interfaced with Agilent Chemstation software and a Syscon REX-C100 air temperature controller (RKC) with a temperature maintained at 25 °C. The 50 mm × 2.1 mm analytical column was packed with a Bidentate C18 stationary phase (BDC18, MicroSolv Tech. Corp.), which had a particle size of 4 µm and a pore size of 100 Å. The final mobile phase obtained after method development used a gradient: Solvent A was premixed 90% deionized water–10% acetonitrile–0.1% formic acid (v/v) and solvent B was acetonitrile with 0.1% formic acid (v/v). The gradient program held at 100% A for 2 min, decreased linearly to 50% A in 7 min, and increased linearly back to 100% A in 1 min. The post time was 5 min. The flow rate was 0.3 mL/min and the injection volume was 2.0 µL.
LC–MS: An Agilent 1200SL Series LC system, including degasser, binary pump, temperature-controlled autosampler, and temperature-controlled column compartment was used. The mass spectrometer system was an Agilent Model 6220 MSD TOF with a dual sprayer electrospray source (ESI). The 75 mm × 4.6 mm analytical column was packed with a BDC18 stationary phase that had a particle diameter of 4 µm and a pore size of 100 Å. The final mobile phase obtained after method development used a gradient: Solvent A was deionized water with 0.1% formic acid (v/v) and solvent B was acetonitrile with 0.1% formic acid (v/v). The gradient program held at 90% A for 2 min, decreased linearly to 40% A in 16 min, and increased linearly back to 90% A in 2 min. The flow rate was 0.4 mL/min and the injection volume was 1.0 µL. Using the positive ionization mode, trans-resveratrol was detected as the [M + H]+ ion (m/z = 229.0859) in the corresponding EIC.
Sample Preparation:
Calibration Curve Solutions: A stock solution of transâresveratrol was prepared by weighing 50.0 mg of the reference standard on a Sartorius Handy H51 analytical balance. The material was quantitatively transferred to a 50-mL amber volumetric flask containing a portion of 50% deionized water–50% acetonitrile. It was sonicated for 10 min using a VWR Symphony ultrasonic bath and then diluted to mark. After mixing, aliquots were transferred to amber volumetric flasks using a calibrated SGE eVol automatic delivery pipette and diluted to mark to obtain concentrations of 0.5, 1.0, 2.0, 5.0, and 10.0 ppm with a diluent of 95% deionized water–5% acetonitrile. Aliquots of the standard solutions were then transferred to 2 mL amber glass autosampler vials (MicroSolv Tech. Corp.). These solutions were then used to produce calibration curve data from HPLC–UV analyses.
Microextraction by Packed Sorbent Percent Recovery Samples: The contents of a capsule containing 100 mg of trans-resveratrol were transferred to a 100-mL amber volumetric flask, and a portion of 50% deionized water–50% acetonitrile diluent was added. The flask was sonicated for 10 min and then diluted to the mark. After mixing, a portion was filtered with a 0.22-µm nylon syringe filter (MicroSolv Tech. Corp.). The theoretical concentration of trans-resveratrol in the stock solution filtrate was 1000 ppm. A 1.00-mL aliquot of the filtrate was transferred to a 100-mL amber volumetric flask and was then diluted to the mark with 95% deionized water–5% acetonitrile and mixed thoroughly. This diluted solution had a trans-resveratrol concentration of 10.0 ppm.
A syringe needle with a C18 sorbent cartridge, either silica hydride based (50 µm, 60 Å, synthesized in house) or type B silica based (45 µm, 60 Å), was attached to the automatic delivery pipette described and conditioned with 100 µL of isopropanol, 100 µL of methanol (two times), and then 100 µL of deionized water. A 100-µL aliquot of the 10.0 ppm trans-resveratrol solution was loaded onto the sorbent and the liquid was discarded. The extract–discard step was repeated zero to three times depending on the preconcentration factor used (see Table 1 for total aliquot load volumes, final concentrations, and analyte mass load). The sample was eluted from the sorbent into a glass amber autosampler vial (with a glass small volume insert) using a 100-µL aliquot of methanol in each case. The samples were analyzed by HPLC–UV and the peak areas were compared to the calibration curve linear model to obtain percent recovery values.

Red Wine Samples (HPLC–UV): A portion of red wine was filtered with a 0.22-µm nylon syringe filter. A syringe needle with a C18 sorbent cartridge (either silica hydride based or type B silica based) was attached to the automatic delivery pipette described previously and was conditioned with 100 µL of isopropanol, 100 µL of methanol (two times), and then 100 µL of deionized water. A 100-µL aliquot of the filtrate was loaded onto the sorbent and the liquid was discarded. The process was repeated for a twofold increase in concentration. The sample was eluted from the sorbent into a glass amber autosampler vial (with a glass small volume insert) using a 100-µL aliquot of methanol. This sample was then analyzed by HPLC–UV and the peak area was compared to the calibration curve line of best fit to obtain an estimate of concentration and then divided by two to take into account the 2× factor from the preconcentration.
Red Wine Samples (LC–MS): To prepare a transâresveratrol stock solution, 1.00 mg of resveratrol was dissolved in 1.00 mL of a solution containing 50% deionized water–50% acetonitrile–0.1% formic acid. The sample for LC–MS analysis was diluted 1:100 with 50% deionized water–50% acetonitrile–0.1% formic acid (v/v).
A red wine sample was prepared by filtering through a 0.45-μm nylon filter (MicroSolv Technology Corp.) and diluting 1:5 with 50% deionized water–50% acetonitrile–0.1% formic acid (v/v). This sample was analyzed by LC–MS as well as a red wine sample spiked with the reference standard solution.
Results and Discussion
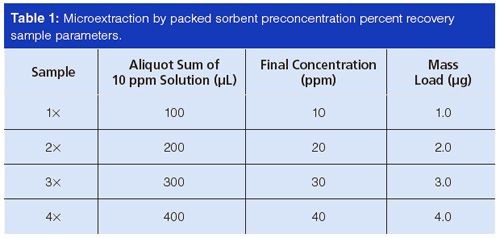
It was important to use amber flasks and vials in this investigation because trans-resveratrol has been reported to degrade upon exposure to sunlight (16). The formation of a degradant means that some of the trans-resveratrol has been used up, skewing quantitative accuracy. Indeed, the effect of amber versus clear vials was studied during method development and degradation was attenuated using the former. Figure 1 shows inter-day overlays of the same 10 ppm transâresveratrol standard solution, with one portion stored in an amber autosampler vial (Figure 1[a]) and another in a clear autosampler vial (Figure 1[b]). A second peak began to appear in both samples, but the amber vial showed less degradation in the studied timescale. The extra peak was significantly more prominent using the clear vial after three days.

Using reference standard solutions, a calibration curve for trans-resveratrol could be constructed on which to base subsequent percent recovery and sample quantitation studies. To optimize sensitivity in terms of the detector, trans-resveratrol’s maximum absorption wavelength of 304 nm was chosen. The data showed good linearity in the studied range of 0.50–10.0 ppm trans-resveratrol (r2 = 0.99992), with a line of best fit equation y = 22.6x – 2.12. Estimates for limit of detection (LOD) and limit of quantitation (LOQ) were also obtained using the signal-to-noise ratio (S/N) of the analyte peak from the 0.5 ppm calibration curve solution (S/N = 91.3). With S/N definitions of 3 for LOD and 10 for LOQ, the values were then estimated at 0.017 ppm and 0.055 ppm, respectively.
To ensure quantitative recovery in the microextraction by packed sorbent samples, various aspects of the technique should be considered and optimized. The loading solvent is an example of one such parameter. Because the retention of resveratrol on the sorbent proceeds via a reversed-phase mechanism, the water content should ideally be as high as possible. In practice, the low solubility of resveratrol in water (24) will require that some acetonitrile be present to allow for complete dissolution. As such, a diluent of 95% deionized
water–5% acetonitrile was chosen. If the water content is too low, quantitative adsorption of resveratrol onto the sorbent may not be achieved, leading to lower recovery.
The choice of the eluting solvent is equally important to ensure that the analyte is quantitatively desorbed from the cartridge and into the autosampler vial. For this reason, the composition of the eluting solvent should consist of at least 60% strong solvent and ideally 100% (25). In reversed-phase, the strong solvent is generally acetonitrile or methanol and the weak solvent is water. Various eluting solvents were studied (see Table 2), and the results were consistent with theory. The 100% deionized water solvent showed the lowest recovery (3.6%) since transâresveratrol is strongly retained on the cartridge under these conditions. When 15% acetonitrile was present, a significant increase to 55.0% recovery was observed. The use of 50% acetonitrile or greater resulted in nearly complete recovery (94.6%), since these are strongly eluting conditions in reversed phase. Of all the solvents investigated, neat methanol produced the highest recovery at 99.8%. Neat acetonitrile was investigated as well, but it resulted in a distortion of peak shape in the HPLC chromatograms on some occasions (data not shown).


A review of the microextraction by packed sorbent technique (17) recommends a washing step between the loading and elution steps to help remove weakly adsorbed sample contaminants, with a highly or completely aqueous solvent being the ideal choice for reversed phase applications. However, this step was omitted in the present investigation for two reasons. First, the sample diluent was 95% aqueous with standard solutions or 100% aqueous with wine samples, so aspirating and discarding an extra aliquot of water was deemed unnecessary; removal of contaminants was already accomplished by discarding the sample aliquot. Second, the data from Table 2 described earlier demonstrated that 3.6% of the transâresveratrol was leached from the sorbent using deionized water as the eluting solvent. Hence, a washing step would be expected to lower analyte recovery and was therefore detrimental to accurate quantitation.
Resveratrol is a moderately hydrophobic compound. As such, it can be retained in microextraction by packed sorbent with a C18 sorbent and reversed-phase conditions. An experimental silica hydride-based C18 material was compared to a C18 sorbent based on conventional type B silica. Silica hydride is a unique material that is virtually free of residual silanols (>95% of Si–OH moieties are replaced by Si–H groups) (18). These silanol groups are known to produce undesirable effects in HPLC and potentially in microextraction by packed sorbent as well. The Si–H group is also relatively nonpolar, which may impart differences in hydrophobicity among the two materials. These types of differences could translate into advantages in extraction efficiency or ability to remove sample contaminants. Subsequent studies comparing the two sorbents could determine if either of these were the case.
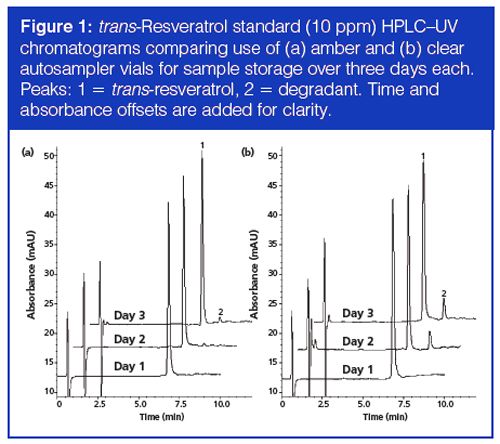
Because the microextraction by packed sorbent cartridge is smaller than the cartridge in an SPE format, the possibility of sample overload should be considered more carefully. If the loading capacity of the sorbent is exceeded, the process may no longer be quantitative, which would defeat the purpose of the technique. Percent recovery studies at increasing concentration factors can help empirically determine whether the technique is quantitative at a given concentration factor. To this end, various trans-resveratrol preconcentration samples were investigated (1–4×) using both sorbents and the percent recoveries were calculated and compared (see Figure 2). The preconcentration can be accomplished by multiple sample load–discard cycles of a given volume followed by an elution of the same volume. The analyst then records the resulting peak areas at each preconcentration level and calculates the percent recoveries. The data demonstrated that only the 2× concentration increase was quantitative while unacceptable deviation from linearity occurred at higher preconcentration levels. In comparing the two types of C18 sorbents, both performed well up to 2×, but neither was able to provide accurate results at the 3× or 4× level. The results suggest that, among other possible explanations, there may be a cumulative effect of a small amount of transâresveratrol leaching from the sorbent bed in multiple extractions, along the same lines as discussed earlier in Table 2. The effect would become more pronounced with increasing numbers of extract–discard cycles, leading to more significant deviation from ideal recovery. This data may also help explain the findings of Gonçalves and Câmara (15), who observed nonquantitative increases in peak area using five and 10 extraction cycles relative to one cycle. For this reason, only the 2× preconcentration factor was used with the subsequent red wine sample.

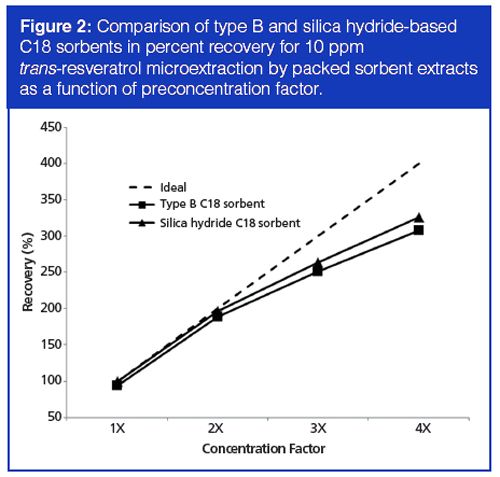
Thus far, the performances of the two sorbents were mostly comparable, with the silica hydride-based sorbent providing slightly higher recovery. However, in the red wine extracts, significant advantages were observed using the silica hydride-based C18 sorbent compared to both the C18 sorbent based on conventional silica and a filtration-only protocol (that is, without microextraction by packed sorbent). Figure 3 compares the chromatograms that were obtained in each case. In Figure 3(a), many peaks are present in the wine sample, especially near the solvent front and in the region where trans-resveratrol is eluted. Because of coelution phenomena from these latter interferences, no trans-resveratrol peak was detected. Use of a type B silica-based C18 sorbent was effective in removing some of these peaks (Figure 3[b]), but still no trans-resveratrol peak was observed. Only with the use of the silica hydride-based sorbent (Figure 3[c]) were enough matrix interferences removed from the extract to detect the analyte peak.

The peak for trans-resveratrol from the 2× red wine microextraction by packed sorbent extract using the C18 silica hydride sorbent could then be correlated with the calibration curve to obtain a concentration of 0.64 ppm in the final sample. Accounting for the twofold increase in concentration via microextraction by packed sorbent, the original concentration of trans-resveratrol in the wine sample was then calculated to be 0.32 ppm. Hence, even though the calibration curve only went down to 0.50 ppm, the analyte could still be made to fall within the studied linear range using the preconcentration strategy. This concentration is within the general range of transâresveratrol in red wine samples reported by Stervbo and colleagues (7).
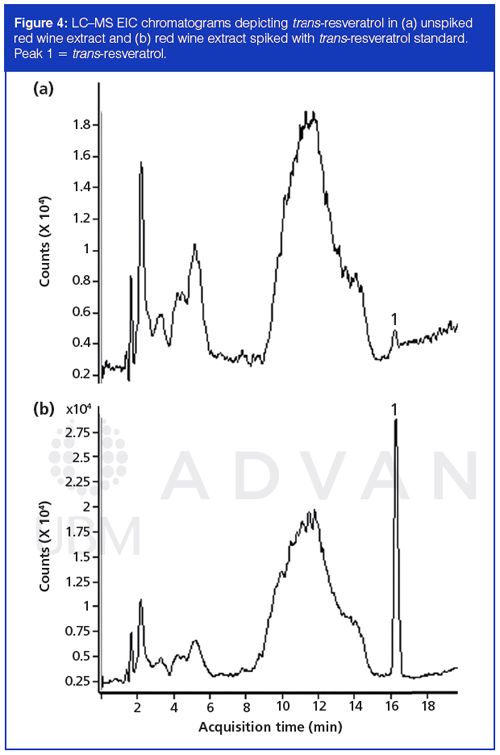
For investigations requiring more sophisticated detection methods, LC–MS was also studied in its application in resveratrol analysis. Figure 4 shows unspiked and spiked red wine extracts using LC–MS without microextraction by packed sorbent treatment. Because of the specificity of MS detection through the use of EICs, a trans-resveratrol peak could be detected using only filtration and dilution in the sample preparation (Figure 4[a]), a feat which could not be achieved in the HPLC–UV investigations. The spiked sample showed that the correct peak assignment was made (Figure 4[b]). The data demonstrate that LC–MS may be an alternative approach to microextraction by packed sorbent in addressing the issue of interfering matrix components in red wine samples.

Conclusion
Using quantitative preconcentration microextraction by packed sorbent approaches with a silica hydride-based C18 sorbent, trans-resveratrol could be adequately detected and correlated to a linear calibration curve model. The same technique was ineffective when a C18 sorbent based on conventional silica was studied because of the insufficient removal of matrix peaks that were coeluted with trans-resveratrol. Likewise, only using filtration without microextraction by packed sorbent was also shown to be impractical with UV detection. However, trans-resveratrol was detected with LC–MS without microextraction by packed sorbent because of the added specificity and sensitivity afforded by MS. Hence, two viable approaches can be used in the analysis of transâresveratrol in red wine.
Acknowledgements
The authors would like to acknowledge Microsolv Technology Corporation for donation of the analytical columns used in this study and SGE Analytical for preparing the microextraction by packed sorbent cartridges.
References
- V.W. Dolinsky and J.R.B. Dyck, Molecules 19, 14919–14947 (2014).
- M. Jang, L. Cai, G.O. Udeani, K.V. Slowing, C.F. Thomas, W.W. Beecher, H.H.S. Fong, N.R. Farnsworth, A.D. Kinghorn, R.G. Mehta, R.C. Moon, and J.M. Pezzuto, Science275, 218–220 (1997).
- E. Tellone, A. Galtieri, A. Russo, B. Giardina, and S. Ficarra, Oxid. Med. Cell. Longev. 2015, 1–15 (2014).
- J. Tomé-Carneiro, M. Larrosa, A. González-Sarrías, F.A. TomásâBarberán, M.T. García-Conesa, and J.C. Espín, Curr. Pharm. Des.19, 6064–6093 (2013).
- C. Andres-Lacueva, M. Urpi-Sarda, R. Zamora-Ros, and R.M. Lamuela-Raventos, in Plant Phenolics and Human Health: Biochemistry, Nutrition, and Pharmacology, C.G. Fraga, Ed. (John Wiley & Sons, New York, USA, 2010), chapter 13, pp. 265–297.
- J.M. Smoliga and O. Blanchard, Molecules19, 17154–17172 (2014).
- U. Stervbo, O. Vang, and C. Bonnesen, Food Chem.101, 449–457 (2007).
- L. Mark, M.S.P. Nikfardjam, P. Avar, and R. Ohmacht, J. Chromatogr. Sci.43, 445–449 (2005).
- C. Silva, C. Cavaco, R. Perestrelo, J. Pereira, and J.S. Câmara, Metabolites4, 71–97 (2014).
- E. Candish, A. Gooley, H.-J. Wirth, P.A. Dawes, R.A. Shellie, and E.F. Hilder, J. Sep. Sci. 35, 2399–2406 (2012).
- M. Abdel-Rehim, J. Chromatogr. B 801, 317–321 (2004).
- J.E. Young, J.J. Pesek, and M.T. Matyska, LCGC North Am.33(3), 192–199 (2015).
- A. Dang, J.J. Pesek, and M.T. Matyska, Food Chem. 141, 4226–4230 (2013).
- Y. Nolvachai, C. Kulsing, R.I. Boysen, M.T. Matyska, J.J. Pesek, P.J. Marriott, and M.T.W. Hearn, Food Chem.174, 434–439 (2015).
- J. Gonçalves and J.S. Câmara, J. Sep. Sci.34, 2376–2384 (2011).
- T.S. Figueiras, M.T. Neves-Petersen, and S.B. Petersen, J. Fluoresc.21, 1897–906 (2011).
- M. Abdel-Rehim, J. Chromatogr. A 1217, 2569–2580 (2010).
- J.J. Pesek and M.T. Matyska, J. Sep. Sci.32, 3999–4011 (2009).
Joshua E. Young is with MicroSolv Technology Corporation in Eatontown, New Jersey, USA.
Michael V. Lim, Joshua Topete, Hao Hang, Marion Gahol, Joseph J. Pesek, and Maria T. Matyska are with San José State University in San José, California, USA.













Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.





