The Ideal Chromatography Data System for a Regulated Laboratory, Part IV: Assuring Regulatory Compliance
LCGC North America
The first three articles in this series discussed where and how a CDS fits into a regulated laboratory, the overall requirements for the architecture of a future system, and additional items to enable effective electronic ways of working. The final part of this series looks at regulatory compliance of a future system as well as a summary of the 15 recommendations made in this series.
The first three articles in this series discussed where and how a chromatography data system (CDS) fits into a regulated laboratory, the overall requirements for the architecture of a future system, and additional items to enable effective electronic ways of working. The final part of this series looks at regulatory compliance of a future system and provides a summary of the 15 recommendations made in this series.
In the first article in this series (1) we looked at the role of the laboratory and discussed the concept of the analytical factory together with the controllable and uncontrolled factors influencing the analytical process. In addition, we looked at the requirements for ensuring data integrity throughout the analytical process. We began the second installment (2) by defining the overall system architecture for a compliant chromatography data system (CDS) in a regulated laboratory in more detail. In the third part (3), we described the new functions required to create fully electronic processes and workflows that should be incorporated into a future CDS to improve efficiency and effectiveness. In this, the last part of the series, we look at regulatory compliance features that must be present in any CDS for trustworthy and reliable electronic records and electronic signatures, thereby ensuring data integrity. To complete this series, we summarize all 15 recommendations made, to describe what the future CDS should look like.
Where Are We Now?
Although chromatography data systems operating in regulated laboratories have basic controls for regulatory compliance there is still a lot that is driven by paper, such as system configuration and instrument qualification. The latter is particularly the case, as suppliers use their service personnel to deliver qualification services, but provide reams of paper for them to fill in for their customers to review. Mistakes, especially by the service personnel, abound as the authors have found when reviewing such documents when advising clients. Moving to an electronic process will eliminate many of these problems and allow fast, electronic review by the laboratory staff. Other areas for compliance improvement include increased data integrity features, improving audit trail content and review, as well as handling unattended working.
Where Do We Want to Be?
From the regulatory perspective a CDS operating in a GXP (good manufacturing, laboratory, or clinical practice) regulated environment should be capable of the following functions:
- documenting the software and instrument configurations of the system,
- automated instrument qualification,
- securing metadata and ensuring data integrity,
- enhanced audit trail functionality to meet current regulatory requirements, and
- compliance control for unattended working.
Each of these areas is discussed in turn in the sections that follow.
Requirement 1: Documenting Configuration Settings
A CDS consists of configurable software that is good automated manufacturing practice (GAMP) Software Category 4 (4), and when used in a regulated laboratory, the system must be validated. One area that needs to be documented is the configuration of the system. This consists of two parts: the first is the software and the second is the overall instrument configuration. Typically, the software settings that need to be configured to meet the business and regulatory needs of a laboratory or organization are definition of user types and the corresponding access privileges, password length and complexity, use of electronic signatures, and electronic records protection. Currently few, if any, chromatography data systems allow a user to document these settings without resorting to a paper-based process. Because the data are contained within the system, would it not make sense to have a function that performed this automatically? Incorporating a search function could allow the system to document the changes over time.
Similarly, the configuration of data servers and chromatographs attached to the CDS should also be available to be documented via the software rather than requiring documentation outside of the system as paper records.
Requirement 2: Automated Instrument Qualification
As noted above, execution of operational qualification protocols is traditionally performed manually with the attendant issues of incomplete signing and dating of all appropriate sections. In addition, the documentation review by the laboratory staff may take time and the engineer may be off-site before errors are found. What we envisage is that the operational qualification protocol for each instrument together with the predefined or user-defined acceptance criteria will be available in the CDS and each protocol will be preapproved by electronic signature before execution.
A service engineer or third-party agent will have limited access to the data system to execute the operational qualification (OQ), gather results electronically, where necessary entering data manually, and document and resolve any discrepancies. The CDS must identify the individual carrying out the work via the log-on credentials. Unless the OQ is reviewed and approved by laboratory staff the instrument cannot be used for regulated work; thus there is a driver to ensure timely review and approval of the data and results versus acceptance criteria.
Based on a user-defined period, the time for the next OQ will be set in the CDS and reminders will be sent before expiry to the instrument owner or the person responsible for instrument qualification. If required, a user-defined grace period can be specified in the system after which the instrument would become unavailable for use if an OQ had not been performed.
The automated instrument qualification procedure is defined by the vendor, but the scientific soundness is attested to by the user. Therefore the procedures and qualification standards employed must be defendable both in terms of good science and traceability to a national or international standard. Currently some vendor practices do not meet these requirements in the second respect. Hence, it would be ideal if the vendor provided the automated tools, but allowed the user to configure the reference materials used to determine criteria such as wavelength accuracy, response linearity, and resolution. However, any change in the acceptance criteria would have to be scientifically sound and justified within the system.
Requirement 3: Securing Metadata for Ensuring Data Integrity
One of the reasons for writing this series is the issue of data integrity in falsification cases found during European Union (EU) and Food and Drug Administration (FDA) inspections (5). The data files generated by any CDS are checksummed to detect and prevent tampering with them. However, examination of data falsification warning letters shows that the main thrust of falsification attempts are manual changes of factors, purities, sample weights, and integration parameters. Therefore, of necessity, data integrity and the associated audit trail entries must cover any changes made to the contextual metadata generated during any chromatographic analysis. This is vitally important as a value of 7.5 is useless without the context of the measurement with respect to units, composition, analysts, instrument, column, lot number, analytical method, and so forth. These contextual metadata are also essential for long-term retention and archiving.
Therefore, in the new-generation CDS it is essential to ensure that only changes to sequence, instrument control, data acquisition, and processing files can only be made by authorized users. This is particularly important for integration parameters. The overall requirements in the data integrity life cycle can be seen in Figure 5, which we presented and discussed in part I of this article series (1).
Requirement 4: Improved Audit Trail Review
Although all CDS applications used in regulated laboratories have audit trails, they are not adequate to meet today’s regulations in an effective way. The key requirement is for audit trail entries to be reviewed by a second person (6–9). According to Annex 11 (6), data entries that have been modified or deleted need to be tracked. This applies to both the chromatography data files, for example, manual intervention in the integration of peaks as well as monitoring changes to the associated metadata used by the run such as sequence file and instrument, acquisition and processing methods, and so forth. The design of the audit trails needs to be smarter as well-it is not the sole purpose in a reviewer’s life to trawl through hundreds of audit trail entries as a chromatographic version of Indiana Jones. CDS suppliers need to define an audit trail dashboard that covers all data and metadata in a run and present this as a traffic light. Traffic lights would work on the principles that green shows where no operator changes or deletions have been made to data, yellow shows where there have been modifications, and red show any data deletions (if allowed by access privileges). This would allow a second person to review by exception only those entries in yellow or red. An alternative approach could be a function that automatically identified modifications or deletions then notified a supervisor or administrator at the start of the second-person review.
The new function also needs to record that the audit trail has been reviewed by a second individual and no action was needed (all green entries) or modifications have been reviewed and that they are acceptable and within the laboratory’s procedures. Also, the ability to set review frequencies on each audit trail (policies, if you prefer) would be a good feature as the function could generate a reminder when a review interval is reached.
For the future CDS, we also need a function that tracks the export of data to other systems via audit trail entries. Many stand-alone systems permit a person to run an assay several times, then pick their favorite run and forward to a laboratory information management system (LIMS). These systems do not track the forwarded runs, so there is no way to quickly identify raw data that is still not included in some test record (at least justified in the CDS as to the rationale for the selection of the data forwarded). Agreeing on injection naming conventions-linked to CDS functionality would help here, along with a simple secure injection sequence log, where appropriate justification is provided as to why each injection in the sequence is performed. Although this may seem draconian, it could make instances of incomplete data, or where the wrong naming convention has been applied, visible in a second-person review.
After all these audit trail functions have been validated, a laboratory can ensure that many second-person reviews can be made speedier and much more efficient.
Requirement 5: Compliance Control in Unattended Analysis
One of the issues with current networked chromatography data systems is that if a run is started and a user goes home how can any changes be made to the run by an authorized user? The assumption made by most, if not all, chromatography data systems is that the user logged in at the start of the run is the same one that makes any subsequent changes, which may not be the case. There needs to be a function, linked to the audit trail, that if an authorized user needs to access a run when the initiating user is not available they can log on and make changes that are attributed to the new user’s identity rather than the originating user.
Regulatory Enhancement Summary
In this article, we have looked at five areas that we believe will bring better regulatory compliance when using a CDS in a GXP laboratory. The ability to document configuration settings quickly and effectively will be useful in initial validation of a CDS, audits, and inspections as well as periodic reviews. Automated electronic qualification of instruments should be the norm rather than slow and error prone execution of paper protocols. Securing metadata in combination with effective audit trails are key compliance features. These additions, along with a documented review of key audit trail entries by exception during the second-person review, are essential productivity and compliance enhancements of any CDS while maintaining compliance with the regulations. Finally, the ability to secure a system during a long analytical run but also have another user access the system and make authorized adjustments under their own name is a key compliance requirement that is not addressed currently.
Bringing It All Together
In this series of articles, we presented 15 areas for improving a CDS for operating in a regulated environment and in this final section we collate and present them in two diagrams.
Figure 1 presents the high-level view of a future networked CDS system where data are stored in a database. The system is interfaced in the laboratory to the chromatographs, but also to an analytical balance to avoid manual transcription of sample weights. Acquired data and metadata are stored in open file formats to allow long-term record retention. The CDS is also interfaced with other informatics applications such as a LIMS or electronic laboratory notebook (ELN) and a deviation management application. Responsibilities for the system are also outlined in Figure 1, with the laboratory staff who are responsible for analytical aspects of the application and IT staff who are responsible for the configuration of the application, user account management, and backup. Data must be acquired, processed, and stored using open file formats for long term retention and interoperability.


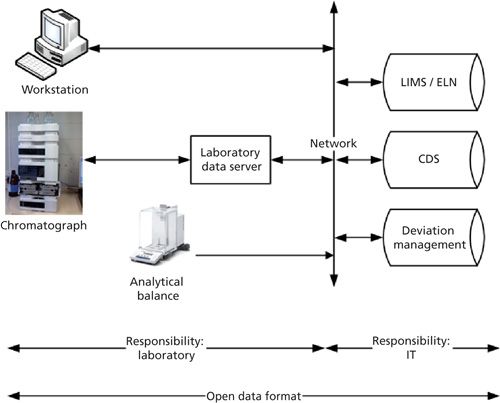
Figure 1: Overall CDS system architecture, informatics connectivity, and responsibilities.
The working of a future CDS in a regulated environment is shown in Figure 2. This figure is based on the overall process flow used in Figures 3 and 5 from part I of this series (1). Under this we have placed four threads: system setup, enhanced compliance, analytical procedures, and electronic working.

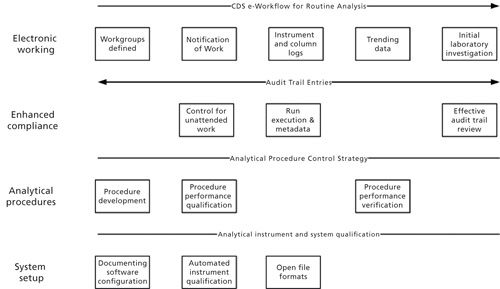
Figure 2: Additional functions and features for a future CDS in a regulated environment.
- System setup covering documentation of system configuration, electronic qualification protocols and their execution by the CDS, and open file formats for the data files and the contextual metadata
- Enhanced compliance features for a new system include compliance control for unattended operation of instruments, means of securing the contextual metadata of an analysis, and effective audit trails to enhance data integrity and second-person data review by exception.
- Analytical procedures covering the spectrum from procedure development, qualification (validation), and verification upon transfer to another laboratory.
- Electronic working including the set-up of workgroups with notification of work to be performed (either analysis or review of data), electronic instrument and column logs that are completed by the CDS rather than manually, trending of data within and between runs, and a user-defined module for performing the initial stages of a laboratory investigation
Although we show these features and functions as stand-alone items this would not be the case in practice. Take, for example, the development of a procedure and its associated procedure performance qualification, data generated during these stages would input into the trending module for the procedure. The analytical control strategy would define the extent of any change that would be allowed without the need to requalify the method, see the process flow in Figure 2 from part I (1). There are further linkages and interactions between other suggested enhancements shown above.
Summary
In this four-part series we have positioned a CDS or similar informatics solution in terms of a regulated environment. The business process that a CDS automates is envisioned as an analytical factory with controlled and uncontrolled factors. The enhancements suggested in this series are intended to ensure that a future CDS can work electronically in an efficient and effective way to generate data with its integrity ensured. Furthermore, the data and metadata are generated in a format that ensures that they can be retained throughout the record retention period.
The 15 proposed areas for enhancement are the major ones envisaged for a future CDS in the short to medium timeframe. They are not intended to be exhaustive or complete. However, these functions will not appear magically in the next release of your CDS system. To be fair to the suppliers of these applications, users need to demand them as these companies are market-led. If you think that these features will be of use in the future, what are you going to do about it?
Acknowledgments
The authors would like to thank Lorrie Scheussler, Heather Longden, Mark Newton, and Paul Smith for comments and suggestions made during the writing of this series of papers.
References
- R.D. McDowall and C. Burgess, LCGC North Am.33(8), 554–557 (2015).
- R.D. McDowall and C. Burgess, LCGC North Am.33(10), 782–785 (2015).
- R.D. McDowall and C. Burgess, LCGC North Am.33(12), 914–917 (2015).
- ISPE, Good Automated Manufacturing Practice (GAMP) Guide, version 5 (International Society of Pharmaceutical Engineering, Tampa, Florida, 2008).
- R.D. McDowall, LCGC Europe27(9), 486-492 (2014).
- European Commission Health and Consumers Directorate-General, Eudralex: The Rules Governing Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use, Annex 11, Computerised Systems (Brussels, Belgium, 2011).
- R.D. McDowall, LCGC Europe25(2), 88–102 (2012).
- R.D. McDowall, LCGC Europe26(6), 338–343 (2013).
- R.D. McDowall, LCGC Europe26(7), 389–392 (2013).
R.D. McDowall is the director of R.D. McDowall Ltd. Chris Burgess is the managing director of Burgess Analytical Consultancy Ltd. Direct correspondence to: rdmcdowall@btconnect.com









Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.

What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?

Advanced LC–MS Analysis for PFAS Analysis in Eggs
October 11th 2024The European Commission's regulation on maximum levels for certain contaminants in food highlights the need for precise and reliable methods to quantify per- and polyfluoroalkyl substances (PFAS) in various food matrices. This article discusses development and validation of a robust method for analyzing 21 PFAS compounds in chicken eggs using solid-phase extraction (SPE) and liquid chromatography–mass spectrometry (LC–MS).




