Flow, Pressure, and Temperature Calibration: Part II
LCGC North America


In last month's installment of "GC Connections," John Hinshaw discussed how peak retention times depend upon relationships between pressure, flow rate, oven temperature, column dimensions, and stationary phase. This concluding installment of a two-part series discusses the effects that column variability has on isothermal capillary gas chromatography and explores instrument calibration with the goal of maximizing instrument-to-instrument similarity of retention times.
Variations in the oven temperature and carrier-gas pressure influence peak retention times significantly enough so that differences from column to column and instrument to instrument make method validation a necessity for ensuring consistent results. Instrument-to-instrument variability can be brought under control by implementing a few simple calibration and set-up procedures, and validation is made more reliable as a result. The first part of this series showed that analysts should understand the effects of performing the same analysis on different gas chromatography (GC) systems on the variability of their results, in particular on retention times (1). The dependencies of peak retention times on oven temperature and inlet pressure can be large enough to cause significant deviations of 15 s or more between instruments when the individual oven temperatures differ by only 1 °C or the pressures by 1 psi. The scope of these variations depends in turn upon the chromatographic conditions, the column, and the analytes under examination. Although not a substitute for validation and suitability testing, instrument calibration can help to reduce the normal variability that will be encountered when working with multiple instruments and multiple columns.
Column Effects
There are three main column variables that affect retention times: dimensional variations such as inner diameter and length; stationary phase variations both in the chemistry and the film thickness; and aging effects due to gradual contamination with sample residue as well as phase loss due to overheating. This month's column addresses some of the issues related to dimensional variations.
I vividly recall spending many long nights in the graduate school lab drawing out borosilicate glass columns on a cantankerous machine that would have made cartoonist Rube Goldberg proud (see www.rube-goldberg.com). If I was lucky enough to obtain a single 10-m long piece of coiled tubing, I then was faced with the tasks of coating the column with a stationary phase that I had synthesized from scratch and installing it intact into the gas chromatograph's oven. I made no pretense of duplicating any of these handmade columns and I don't know what the tolerance levels were on their inner diameters or film thicknesses. Fortunately, this had no bearing on my work. However, it did leave me with an appreciation of the technology that goes into producing capillary GC columns.
Two studies published in the 1970s recorded the state of the art of commercial glass capillary GC column production at the time (2,3). A statistical evaluation of the data published in the two papers (4) reveals retention-factor standard deviations of 5.9% for 16 methylsilicone columns, 11.4% for 7 phenylmethylsilicone columns, and 30.4% for 9 Carbowax 20M columns (2). In the second report, the authors measured the relative retention of several peak pairs and found, not surprisingly, much smaller standard deviations: 0.28-0.37% for methylsilicone and 0.32% for Carbowax 20M. Nonpolar columns prepared with twice the stationary phase film thickness had even smaller column-to-column relative retention variations (3). The variability of the column inner diameter does not affect retention factors under the isothermal conditions used, and its range was given as dc = 270 ± 20 μm (2).
Today, analysts rely upon column manufacturers to produce a consistent product from fused-silica tubing. Advances in tubing production and chemical treatment, stationary phase synthesis, column coating, and conditioning have greatly reduced the variability in column dimensions and retention as well as yielding much lower bleed levels, higher stability, and longer life. It would be very interesting to see similar data on populations of modern capillary columns that compares them with the older studies.
Gas chromatographers who want to ensure the best consistency from column to column should choose one manufacturer as their column source for each specific analytical method. There are no technical reasons to select or eliminate any particular manufacturer, but columns from the same company will be much more self-consistent than would be the case for columns from different manufacturers with the same nominal dimensions and stationary phase types. Of course, some column companies' proprietary stationary phases and column chemistry might be better suited for certain applications. Careful evaluation of multiple examples of a specific column is always a good idea before committing to any particular choice.
Batch-to-batch variations in stationary phase chemistry are important because they affect peak retention directly, but in this article, I will assume that the columns all are the same in this respect. Working within the tolerances of the old column study, which can be taken as larger than the absolute maximum range that would be encountered today, what then are the effects of variability in column diameter and length on retention times?

 Inner diameter = 200–300 μm; (b) close-up view, inner diameter = 240–260 μm. Column: 25 m x 250 μm; column temperature: 100 °C; pressure drop: 15 psig, column outlet at room pressure. Key: (blue) n-dodecane, K100 °C = 522.1; (green) n-undecane, K100 °C = 270.2; and (red) n-nonane, K100 °C = 70.99.")
Figure 1: Effect of column inner diameter on retention times. (a) Inner diameter = 200–300 μm; (b) close-up view, inner diameter = 240–260 μm. Column: 25 m x 250 μm; column temperature: 100 °C; pressure drop: 15 psig, column outlet at room pressure. Key: (blue) n-dodecane, K100 °C = 522.1; (green) n-undecane, K100 °C = 270.2; and (red) n-nonane, K100 °C = 70.99.
Column Inner Diameter
The column diameter affects both the average carrier gas linear velocity and the retention factor, given a constant stationary phase film thickness. The linear velocity will decrease as the square of the inner diameter decreases, as equation 1 shows.

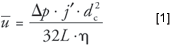
We can compare the effect of different column inner diameters on velocity at constant column pressure, length, and temperature as follows:

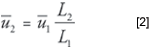
This type of square-law relationship predicts a strong dependence of the linear velocity, and thus retention times, on the column diameter.

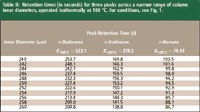
Table I: Retention times (in seconds) for three peaks across a wide range of column inner diameters,operated isothermally at 100 °C. For conditions, see Fig. 1.

Table I shows the effect of changing the column inner diameter across a relatively wide range — from 200 to 300 μm — on the retention times of the same three example hydrocarbon peaks as used in the first part of this series, and the data are presented graphically in Figure 1a. The effects on retention time are large. Table II gives the same data for a narrower range of inner diameters, from 240 to 260 μm, as might be encountered in practice, and the corresponding plot is shown in Figure 1b. According to this information, to keep retention times within a maximum range of ±15 s, the inner diameter would have to fall within ±6 μm or about ±2.4% of the nominal 250-μm inner diameter for the longest retained peak shown here; within ±10 μm for the middle peak; and within ±20 μm for the earliest eluted peak.
As peak retention increases, the variability in inner diameter required to keep peaks within a defined range decreases rapidly. However, with isothermal elution, the widths of the peaks increase with longer retention, and the effect of the variability becomes less significant. This is not the case for temperature-programmed elution, but this topic lies outside of the discussion being presented here.

Table II: Retention times (in seconds) for three peaks across a narrow range of column inner diameters, operated isothermally at 100 °C. For conditions, see Fig. 1.
Thus, it appears that for columns of the same type used for the same isothermal analysis, if the inner diameters from one column to the next lie within less than ±2% of the nominal diameter, then peaks will be eluted within a fairly tight window. Remember, though, that other variables are also at play here. Temperature and pressure variability will add more uncertainty to the retention times.
Column Length
Variations in column length also affect retention times. Differences between column lengths on the order of 1 m or more are not uncommon within a population of initially equal-size columns that have been in use for some time. Removal of a small portion of a column is part of good laboratory practices that call for the use of new inlet and detector ferrules with each installation. Additional lengths can be removed from the column entrance as part of column reconditioning in order to remove nonvolatile sample residues that have accumulated at the beginnings of columns.
The effects of varying column lengths depend upon how the analyst sets up the columns. One approach would be to choose the same pressure drop for all columns of a particular type. In this case, retention times will vary with the square of the column length, as shown in equation 3, which was obtained by combining equations 1, 2, and 3 from the first installment of this column series (1):

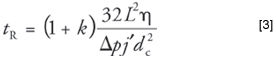
This is not a desirable situation. The peak with partition coefficient K = 522 at 100 °C (n-dodecane), which is eluted in 227 s on a 25-m column with a 15.0-psig pressure drop, would shift to 210 s on a 24-m column with the same pressure drop. The effect on later-eluted peaks would be even larger.
If, on the other hand, the carrier-gas linear velocity were set the same for the 24-m column as the 25-m column, by adjusting the inlet pressure downward slightly to 14.4 psig for the shorter column, then the same peak would shift by about half as much, to around 218 s. Keeping the retention time the same for both column lengths would require a further decrease in the shorter column's pressure drop to around 13.8 psig. This pressure corresponds to an average carrier-gas linear velocity that is exactly the ratio of the two columns' lengths times the original velocity. In this case, that's 24.0/25.0 × 34.0 = 32.64 cm/s. The other peaks follow suit in this case and have the same retention times on the shorter column under these lowered inlet pressure conditions as they do on the longer column at 15.0 psig.
Setting up by linear velocity: In practice, it's fairly easy to set up a column in this manner. First, measure the approximate length of the column by counting the turns (include any fractional first or last turn) and multiplying by the average length of a single turn, as in equation 4:


where tc is the turns count and dh is the nominal column helical coil diameter. Use a value of dh that's close to the apparent average value for the coils of the column.
Next, calculate the ratio of the length of the column at hand to the nominal column length, for which the method conditions were developed. Third, multiply by the desired average carrier gas linear velocity for the method to obtain the linear velocity goal for the specific column being installed, as shown in equation 5:


Finally, as part of verifying the setup, establish the operating or initial column temperature and then adjust the inlet pressure as required to produce the calculated velocity.
For GC systems with electronic pressure control (EPC), the EPC system will calculate and set the correct pressure for the desired velocity if the measured column length is entered first. However, slight variations in the column diameter from the nominal diameter can produce a slight error in this step. The operator can approximate the apparent column diameter at this point by comparing the measured carrier gas linear velocity to the desired value as entered into the EPC system. Because, according to equation 2, the ratio of the velocities is equal to the square of the ratio of the diameters, a corrected diameter can be calculated in this manner:

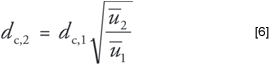
For the example given above, if the desired velocity was 32.64 cm/s for the 24 m × 250 μm column, but the observed velocity was 34.9 cm/s, then the corrected column inner diameter would be:

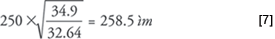
Upon entering this corrected inner diameter into the EPC column configuration, the electronic pressure controller will adjust the pressure downward sufficiently to produce the desired average carrier-gas linear velocity. Due to the variations in inlet pressures from instrument-to-instrument, however, this procedure should be repeated whenever a column is set up.
Temperature and Pressure Calibration
With a better understanding of the effects of column variability on retention times, we are in a position to examine the requirements and effects of temperature and pressure calibration in laboratories that utilize multiple gas chromatographs.
What to expect: Given that small changes in temperature or pressure can shift retention times significantly, what should gas chromatographers expect from their instrumentation? How much pressure and temperature variation is normal between gas chromatographs that are operating within the manufacturer's specifications? I read through a number of brochures, specification sheets, operator's manuals, and service manuals. I found that pressure and temperature tolerances vary somewhat by manufacturer and for some items, there was little or no information. Most GC systems produced in the past 10-15 years include provisions for oven temperature, carrier pressure, and flow calibration via the instruments' keyboard-display and firmware. Thus, gas chromatographers can bring these variables under some degree of control.
Temperature: In my experience, average oven temperatures vary between instruments by as much as ±2 °C or slightly more. When left uncalibrated, larger deviations can be expected between different models than between the same model gas chromatographs. The apparent degree of variation also depends upon how the temperature is measured. All GC ovens exhibit temperature gradients between the internal temperature sensor, where the temperature is measured, and other locations within the oven. The degree of temperature gradients depends upon many factors, such as the oven temperature setpoint; whether the cooling flap or door is open or shut; the condition of the door closure and insulation; the temperatures of the inlets and detectors; and any other material, such as columns, valves, and other accessories, that affect heat and air flow. Gradients of as much as 2-4 °C across the oven are not uncommon even under the best circumstances.
One very important consideration is the distinction between the oven temperature setpoint as displayed on the GC, calibrated or not, and the actual temperatures along the column. Columns are not located at the temperature sensor, which measures only a single point anyway. As peaks move through the column, they circle around with the column tubing and run through slightly hotter and slightly cooler temperature areas. Upon elution, they have experienced an average temperature that is a composite of the temperature at any defined point in the oven. This is the normal situation and these slight temperature variations don't affect peak shapes or resolution significantly.
Placing a column too close to the oven wall will increase this effect, because the coolest areas in the oven tend to be nearest the walls. Conversely, the hottest areas often are near the inlets and detectors. Shifting a column's position from the front of the oven to the back can have a noticeable effect on retention times as well. Thus, for the best consistency, it is wise to install columns close to the central axis of the oven and always either in the front or the back position as dictated by the inlet-detector configuration and the methodology. As long as the overall thermal environment is consistent, the retention times will be as well.
Pressure: Carrier-gas inlet pressures are controlled either by electronic pressure controllers or by manual regulators, which may or may not have electronic pressure gauges. For capillary columns with inner diameters less than 530 μm, a pressure-controlled split-splitless inlet system is the most common. Even for EPC in the constant-flow mode, with this type of inlet, the GC system actually controls the inlet pressure and sets it as required to maintain the desired flow rate, using relationships derived from equations 3-5 from the first installment of this column (1). Wide-bore columns of 530 μm i.d. and up can use a true flow-controlled carrier source instead.
If a Bourdon-type mechanical pressure gauge is in use, then there is little realistic need to calibrate it because it is inherently inaccurate and nonlinear, compared to electronic transducers. In such cases, chromatographers should rely on the gauge as an approximate pressure indicator and instead use carrier-gas average linear velocity measurements for column setup purposes. I also have seen a digital electronic pressure device with an attached syringe needle, which gives a fairly accurate reading when inserted into an inlet.
Most of the electronic pressure transducers for EPC systems are specified to deliver accuracies of ±2% of their full-scale reading. For a 0-100 psig transducer, that's about ±2 psig, while for a 0-30 psig transducer, it's more like ±0.6 psig. These errors show up as deviations from a perfectly linear relationship between the actual pressure and the readout, and according to our calculations, they are large enough to cause noticeable retention shifts from one instrument to another. The solid-state pressure transducers cannot be calibrated by the instrument for their nonlinear full-scale errors, but all of these GC systems support a zero offset adjustment, which can be as large as an additional ±0.5 psig or so.
Calibration
Properly performed, temperature calibration and inlet pressure zero-compensation can help tremendously in attaining more consistent instrument-to-instrument results. However, it also is necessary to consider carefully other variables such as column dimensional variations and positions inside the column oven.
Oven temperature: A precision temperature measuring device and appropriate probe are necessary for meaningful oven calibration. The probe and meter combination should be capable of 0.2 °C or better resolution. If consistency of results between instruments that will be calibrated with different thermometers is important, then the thermometers should all be calibrated to NIST standards by their manufacturers. For single locations, a single thermometer should work well enough with a simple ice-bath reference.
It is very important to place an external temperature probe consistently when measuring oven temperatures for calibration purposes. Bearing in mind that in any case the calibration will only reflect a single-point temperature, the best placement is as close as possible to the instrument's temperature sensor. If variations between different instrument models are a concern, then slightly better results might be obtained by positioning the temperature probe close to the center of the oven in the area where the column will be located. In any case, the probe should never be placed close to the oven walls or directly in a line of sight with the oven heater coils. Be sure that the probe cable does not interfere with the gas chromatograph's oven door and that introducing the probe does not create an extra air leak from the outside.
See the user's manual or service manual for details on exactly how to calibrate the temperature for a specific instrument. In general, operate the GC system at a temperature in the middle of the method operating range, or at 100 °C. A well-controlled room temperature also helps attain more consistent oven temperature calibration.
Let the instrument stabilize for at least 1 h, and then access the instrument calibration routine. Compare the resulting high-resolution reading with the probe reading, and enter a corresponding temperature offset value on the keypad, or in some cases, enter the probe temperature reading itself as instructed in the manual. Allow some time for the new temperature level to settle in, and then verify that the probe and oven now are consistent to within a few tenths of a degree. Make a note of the temperature probe make and model, its position in the oven, how it was calibrated, and the offset value in effect after calibration.
Inlet pressure: As mentioned earlier, pressure calibration is not practical to better than ±2% of the full-scale reading. If desired, however, an external digital pressure measurement device can be used to obtain an independent pressure reading at or close to a single setpoint. Such readings will be a valid indication of the relative pressures in multiple instruments for the purposes of setting up a method that uses constant inlet pressure. However, if the method calls out any type of pressure programming, including constant flow mode with temperature programming, then these readings will only set the initial pressures and will have little bearing on subsequent control changes. In the case of a mechanical pressure gauge, then an external digital transducer becomes a very valuable tool.
However, the pressure and related flow transducers in a GC should be zeroed at least every three months, as well as whenever the instrument is moved or serviced. If the pressure readout is not 0.0 when the pressure is off and no column is attached, then the associated transducer should be zeroed.
To zero the transducers, first cool down the column oven, then turn the carrier gas off or set the pressure to zero and either disconnect the columns or remove the septum nuts from the inlets. Most EPC-equipped instrument models also monitor the incoming carrier-gas supply pressure, so disconnect the carrier-gas supply at the instrument bulkhead, being careful to cap off the supply tubing to protect carrier-gas filters from air incursion.
Allow at least a half hour for the instrument to warm up, if it is not already warmed, then select the pressure transducer zeroing portion of the keyboard-display user interface and execute the zeroing procedure according to the user manual. Finally, reconnect the supply lines and establish a low pressure for long enough to purge air from the system before reconnecting the columns or replacing the inlet septum nuts. This also would be a good time to service the inlets if necessary.
Conclusion
Some variability in results obtained on different instruments with different columns is always to be expected. Analysts can minimize instrument-to-instrument retention time variability by calibrating the oven temperature, installing the column in the same oven location, and setting the carrier-gas average linear velocity to compensate for slight column-to-column variations in length and inner diameter. Good column maintenance practices also will help establish better repeatability. Although not discussed in detail in this article, with careful thermal calibration, dimensionless retention measurements such as the retention factor, relative retentions, and retention indices inherently rationalize interinstrument variations and intercolumn dimensional variations and effectively make such results more comparable.
References
(1) J.V. Hinshaw, LCGC 22(12), 1160 (2004).
(2) M.J. Hartigan and L.S. Ettre, J. Chromatogr. 119, 187-206 (1976).
(3) M.J. Hartigan, K. Billeb, and L.S. Ettre, Chromatographia 10, 571-579 (1977).
(4) L.S. Ettre, personal communication, November 2004.
John V. Hinshaw "GC Connections" editor John V. Hinshaw is senior staff engineer at Serveron Corp., Hillsboro, Oregon, and a member of LCGC's editorial advisory board. Direct correspondence about this column to "GC Connections," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com. For an ongoing discussion of GC issues with John Hinshaw and other chromatographers, visit the Chromatography Forum discussion group at http://www.chromforum.com




Fundamentals of Benchtop GC–MS Data Analysis and Terminology
April 5th 2025In this installment, we will review the fundamental terminology and data analysis principles in benchtop GC–MS. We will compare the three modes of analysis—full scan, extracted ion chromatograms, and selected ion monitoring—and see how each is used for quantitative and quantitative analysis.




From Gas to Gas: Fundamentals of Static Headspace Extraction-Gas Chromatography
August 9th 2024In this article, we discuss the fundamentals of headspace extraction, including static versus dynamic extraction, establishing equilibrium in the vial, consequences of the partition coefficient, temperature, pressure, and transfer to the gas chromatograph.


