Field-Flow Fractionation for Biological, Natural, and Synthetic Polymers: Recent Advances and Trends
LCGC Asia Pacific
A review of the latest trends in field-flow fractionation (FFF) for various types of polymer analysis.
Field-flow fractionation (FFF) is a family of techniques that is increasingly used for separating and characterizing macromolecules. This review discusses recent advances in the characterization of biological, natural, and synthetic polymers. Applications of FFF are contrasted with size-exclusion chromatography to illustrate practical considerations when characterizing macromolecules. The use of different FFF fields allows separations based on size, mass, composition, and architecture. The open channel design and subsequent low shear rate is well suited for analyzing weakly bound complexes, highly branched polymers, high molar mass analytes, and aggregates. Other benefits of FFF that are highlighted in this paper include simplified sample preparation, flexibility in carrier fluid choice, and on-line removal of low-molecular-weight contaminants.
Macromolecules are ubiquitous in many areas of science and technology. Depending on the macromolecule, it is important to analyze properties like size, molar mass (MM), chemical composition, degree of branching, and their respective distributions to understand their behaviour. However, because of the complex nature of polymers, current separation techniques are not always capable of comprehensive analyses. Size-exclusion chromatography (SEC) is widely regarded as the workhorse for polymer characterization, but is limited by high molar mass (HMM) macromolecules, weakly bound complexes and aggregate species, and highly branched polymers. Field-flow fractionation (FFF) is a versatile family of techniques that complements SEC with additional separation capabilities based on analyte size, mass, composition, or architecture depending on the field used (Figure 1).


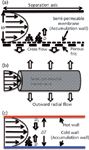
Figure 1: Types of FFF separation. (a) In AF4 a crossflow passes through a semi-permeable membrane and porous frit. (b) In HF5 a cylindrical semipermeable membrane is used and a radial outward flow creates the perpendicular field. (c) In ThFFF a temperature gradient (ÎT) is formed between a hot wall and a cold wall, and sample migrates towards the cold wall because of thermal diffusion (DT).
The open channel FFF design results in a soft separation mechanism that is well suited for analysis of high and ultrahigh MM polymers and samples containing microgel. Some key advantages of FFF over SEC arises from its ability to separate analytes over a broad size range (0.001 to 100 µm) using a single channel, and the absence of column packing, which greatly reduces shear degradation. SEC of protein aggregates often requires the addition of cosolvents or preconditioning of columns to reduce adsorption (1). However, addition of cosolvents may induce aggregation, dissociate aggregates, or cause sample specific adsorption (Figure 2) (2). Preconditioning columns is often practised but not reported in the literature, and even when preconditioning is used poor recoveries and sample specific adsorption have been observed (3). In FFF the ability to use formulation buffer allows separations and measurements under solution conditions that are more representative of actual use. For polymer analysis, the shear degradation and co-elution of small and large analytes observed in SEC for highly branched polymers are attributed to effects caused by the column packing material (4).



Figure 2: An IgG1 recombinant fully humanized monoclonal antibody was analyzed by FFF in two different carrier fluids: (a) 0.1% acetic acid containing 50 mM magnesium chloride and (b) 10 mM phosphate buffer pH 7.1. High molar mass aggregates (peak at ~18.5 min) present in (a) are absent in (b) as a result of weak aggregate interactions stabilized by the magnesium chloride. Adapted and reproduced with permission from B. Demeule, M.J. Lawrence, A.F. Drake, R. Gurny, and T. Arvinte, (2007), Biochim. Biophys. Acta-Proteins. Proteomics 1774, 146-153. © Elsevier.
In practice, FFF offers users additional benefits. Prior to SEC, filtering is often implemented as a sample preparation step to remove large components and help prolong the life of the column. Sample filtering has been shown to remove soluble and insoluble microgels leading to erroneous MM and polydispersity results (Figure 3) (5). Filtering is not required in FFF and soluble polymers and microgels can be simultaneously characterized. Many syntheses require the addition of excess reagents, which may interfere with subsequent product analyses. Such reagents or interfering low MM sample components either elute in the void peak or can be removed on-line through a semi-permeable membrane used in some FFF techniques.


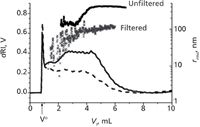
Figure 3: ThFFFâMALSâdRI analysis of unfiltered (solid line, black symbols) and 0.5-µm filtered (dashed line, grey symbols) microgel-containing poly(vinyl acetate). The lines and symbols represent the dRI fractograms and rg, respectively. Significant polymer loss in the filtered sample is evident in the lower MM distribution. Adapted and reproduced with permission from D. Lee and S.K.R. Williams, (2010), J. Chromatogr. A. 1217, 1667-1673. © Elsevier.
Separations in FFF are dependent on the strength of an externally applied field which can be easily adjusted. Therefore, resolution and separation speed are readily controlled without the need to change channels. In addition, the open channel design greatly reduces the chance of contamination and inexpensive membranes can be replaced when contaminated. Finally, FFF is easily coupled on-line with detectors frequently used for SEC analysis, including multi-angle light scattering (MALS), differential refractive index (dRI), and mass spectrometry (MS) detectors. For those interested in FFF, building a simple homemade system requires a FFF channel and standard high performance liquid chromatography (HPLC) components common to many laboratories. The recent advances in FFF over the last three years are highlighted in this review.
Principles of FFF
Separation takes place in a thin, open, ribbon-like channel where carrier fluid transports components down the separation axis of the channel. Frictional drag at the channel walls creates a parabolic flow profile across the channel thickness, w, with the fastest flows in the middle of the channel and the slowest flows near the walls (Figure 1). An external field (flow, thermal, or sedimentation) is applied perpendicular to the separation axis of the channel to drive components towards the accumulation wall. This field-induced transport is counteracted by diffusion of components away from the high concentration region near the accumulation wall. Equilibrium is reached when the two transport processes are balanced and there is no net flux of sample in either direction. The equilibrium position is different for each sample component depending on the magnitude of their interaction with the applied field and their diffusion coefficient. Components of smaller sizes diffuse further into the channel than larger components based on the inverse relationship between diffusion coefficient (D) and hydrodynamic diameter (dh) given by the Stokes-Einstein equation for spherical analytes. The smaller components experience the faster flows further from the accumulation wall and therefore elute before larger components in normal mode FFF. This normal mode elution order is the reverse of that observed for SEC.
The main FFF techniques relevant to this review are asymmetrical flow field-flow fractionation (AF4), hollow fibre flow field-flow fractionation (HF5), and thermal field-flow fractionation (ThFFF). AF4 utilizes a single permeable wall that allows a crossflow to act as the perpendicular field (Figure 1[a]). The permeable wall is composed of a porous frit covered with a semipermeable membrane, the latter acting as the sample accumulation wall. The retention time (tr) for AF4 is given in equation 1:



where η is the carrier fluid viscosity, t0 is the void time, Vc is the crossflow rate, V0 is the void volume, k is Boltzmann's constant, and T is the temperature.
In HF5 a semipermeable hollow fibre membrane is used and an outward radial flow acts as the perpendicular force (Figure 1[b]). The benefits of HF5 over AF4 are lower sample volumes and a potentially disposable channel. Thermal FFF (ThFFF) employs a hot and cold wall to create a temperature difference (ΔT) that subsequently induces thermal diffusion of components towards the cold wall (in most cases) (Figure 1[c]). The retention time is given in equation 2


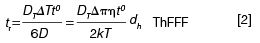
where DT is the thermal diffusion coefficient.
Biopolymers
Biopolymers are a diverse class of macromolecules that includes polypeptides, polynucleotides, and polysaccharides. The versatility of AF4 separation and the characterization of biopolymers is well established. Several review papers and a book focusing on the analysis of biological polymers using FFF have recently been published (6–8).
Characterizing protein—protein and protein—macromolecule complexes is important for understanding the efficacy and functions of proteins. AF4's gentle separation mechanism is well suited to analyze complexes with weak interactions (Figure 4) (8). Current techniques for characterizing protein dissociation constants (Kd), such as surface plasmon resonance (SPR), analytical ultracentrifugation (AUC), and SEC, are limited in their ability to analyze more than two components or to detect weak binding affinities (Kd > µM). Protein—protein binding between a neonatal Fc receptor (FcRn), immunoglobulin (IgG), and human serum albumin (HSA) was recently studied by AF4 (9). FcRn is involved in removing IgG proteins from lysosomal degradation pathways and IgG transportation in the body. AF4 separation of the IgG-FcRn complex allowed for the determination of a relatively low binding affinity (Kd of 3.74 µM). In addition, FcRn, HSA, IgG, and their associated complexes were separated using AF4. By using an internal standard curve, the formation of multi-protein complexes were determined, including a previously unreported protein complex (HSA/FcRn/IgG/FcRn, 303 kDa). The separation of intact, weakly bound protein complexes shows great promise for AF4 studies of protein pharmacokinetics and aggregation kinetics. Analysis of protein aggregates, especially those in the submicron size range, is of particular interest in the development of therapeutic proteins. Development of an AF4 method for separating IgG monomer and submicron IgG aggregates was recently shown by Hawe et al. (3). Better size resolution and recoveries of submicron IgG aggregates were achieved by AF4 compared to SEC.


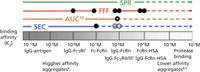
Figure 4: A comparison of FFF to other currently used techniques (analytical ultracentrifugation [AUC]; surface plasmon resonance [SPR]) for proteinâprotein characterization. The open channel FFF design and flow-based separation extends the current ability to detect weak proteinâprotein interactions into the µM binding affinity range. Adapted and reproduced with permission from J. Pollastrini, T.M. Dillon, P. Bondarenko, and R.Y.T. Chou, (2011), Anal. Biochem. 414, 88â98. © Elsevier.
Lipoproteins are assemblies of proteins and lipids that function as carriers for lipids and cholesterols in blood. AF4 has been used to analyze low-density lipoproteins (LDL) and high-density lipoproteins (HDL) (10). LDLs have been associated with an increased risk of coronary artery disease (CAD). In addition to the conventional AF4 channel, a hollow fibre guard channel placed before the AF4 channel was evaluated using serum from healthy patients and CAD patients. The guard channel removed contaminants and improved reproducibility in retention, and fluorescence detection reduced adsorption of serum proteins to the membrane and reduced the amount of serum required for each injection (0.13 µL).
On-line coupling with a variety of detection methods has expanded the breadth of AF4's characterization ability in recent years. The use of MALS, dRI, and quasi-elastic light scattering (QELS) detectors has become more common for characterizing macromolecules. Characterization of dh, MM, radius of gyration (rg), and chemical composition was shown for a PEGylated protein conjugate and its aggregates using on-line AF4–UV-MALS–QELS–dRI, SEC–UV-MALS–QELS–dRI, and matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry (MALDI–TOF-MS) as complementary techniques (11). PEGylated protein, unreacted protein traces, and aggregated species were detected by both AF4 and SEC, with AF4 providing superior size resolution, while MALDI–TOF-MS was unable to detect aggregates. Detection by UV–MALS-QELS–RI enabled chemical composition characterization of PEGylated proteins (1/1 PEG to protein ratio) and allowed identification of aggregates present using different storage buffers.
Interest in characterizing protein complexes by 2D off-line coupling of AF4 with other separation techniques and a variety of detection methods has grown in recent years. Lectin-treated N-linked glycopeptides in serum from lung cancer and healthy patients were separated by AF4 and subsequently analyzed by nanoflow liquid chromatography-electrospray ionization–tandem mass spectrometry (nLC–ESI-MS–MS) (12). Binding of various lectin types to glycoproteins enabled a size-based separation by AF4. Removal of non-lectin bound glycopeptides and size sorting of lectin-glycopeptide complexes during AF4 allowed semi-quantitative analysis and improved identification of biomarkers by nLC–ESI–MS–MS. Similarly, characterizing cholesterols and triglycerides in lipoprotein complexes is also important for understanding their function in the body. Off-line coupling of AF4 and gas chromatography (GC)–MS allowed cholesterols and triglycerides to be profiled from human serum samples, and results showed agreement with the current enzymatic determination methods (13).
The use of FFF has shown promise as a pre-MS separation technique in proteomics analyses. To improve MS detection of poorly soluble proteins, the effect of protein–SDS complexation on protein solubility was examined by HF5 and nLC–MS (14). SDS-denatured serum samples, unfractionated or fractionated, showed improved solubility of the SDS–protein complexes, and allowed for a greater number of proteins to be identified by nLC–MS. Furthermore, the HF5 process was shown to remove low MM (< 30 kDa) components, which subsequently led to lowered background noise in the MS spectrum.
Protein phosphorylation is a post-translational modification which plays an important role in protein regulation and can be used as a biomarker for diseases like cancer. The 2D on-line coupling of an isoelectric focusing (IEF) step and AF4 step prior to nLC–ESI–MS–MS enabled separation of phosphorylated proteins from a proteome sample based on isoelectric point (pI) and dh (15). IEF-AF4 separation was evaluated for unphosphorylated and phosphorylated α-casein. Peptides with higher degrees of phosphorylation eluted in the lower pH channels and at longer AF4 retention times as expected. Relative abundances of phosphorylated protein biomarkers were determined by IEF–AF4 and nLC–ESI–MS-MS for a prostatic cancer line and a normal cell line. In another study, improvements in direct on-line coupling of AF4 with ESI–MS were shown in a small chip-type channel for top-down proteomics that operates in the micro-flow rate regime (Figure 5[a]) (16). The chip-type channel effectively separated carbonic anhydrase (29 kDa) and transferrin (78 kDa) while using much lower, and more ESI–MS compatible, channel flow rates (<12 µL/min) than previous on-line studies (Figure 5[b]) (17,18). Resolution of monomer and aggregate species as well as desalting during AF4 led to higher signal-to-noise for ESI–MS detection (Figure 5[c] and 5[d]). Lipodomic analysis of HDL and LDL from human serum was also shown by chip-type AF4 (19). On-line desalting improved ionization of the HDL and LDL lipids, and in a CAD plasma sample 28 phospholipids, 18 triacylglycerides, and six cholesteryl esters were identified.



Figure 5: (a) A schematic of the chip-type miniaturized AF4 channel interfaced with electrospray ionization mass spectrometry (AF4âESIâMS); (b) Base peak fractogram (BPF) of AF4âESIâMS for the separation of CA and transferrin (Vout/Vc = 0.012/0.49 mL/min; (c) Full scan ESIâMS for peak #2 (transferrin) after AF4 shown in (b); (d) Full scan ESIâMS spectrum of transferrin (0.01 µg/µL) without AF4. Considerably better S/N is observed in the fractionated transferrin as a result of monomer/dimer resolution and contaminant removal during AF4. Adapted and reproduced with permission from K.H. Kim and M.H. Moon, (2011), Anal. Chem. 83, 8652â8658. © American Chemical Society.
Natural Polymers
Starches are macromolecules essential to human beings and are used in a variety of industrial and food applications. The properties of starches (for example, digestion and thickening abilities) are dependent on starch structure, which in turn is dependent on the degree of branching. However, wide size distributions and variation in branching makes characterizing starches difficult (20). AF4 coupled with MALS and dRI detectors has been successfully used to determine starch dh, MM, and rg (21,22). An in-depth review of FFF characterizing food macromolecules has been recently published (23).
Wahlund et al. demonstrated the power of AF4–MALS–RI to rapidly separate amylose and amylopectin in maize, wheat, rice, potato, and tapioca starches (24). Qualitative results for amylose and amylopectin ratios demonstrate the feasibility for relatively fast characterization of starches by AF4 and provide a starting point for more extensive starch analyses. Studies by Juna et al. have examined various starches (waxy maize, tapioca, corn, sago) to better understand AF4 conditions and starch processing parameters (25–30). Changes in size distributions were observed with changes in AF4 conditions. For example, at high cross flow rates, the dh, MM, and rg distributions of tapioca, sago, and corn starch shifted to lower values because of increased retention (or potential degradation of HMM components). The effect of AF4 conditions is therefore important and must be considered for accurate analyses of starches.
Coupling a separation technique with MALS and dRI detectors can provide information on structural and branching characteristics of starches. SEC is the most common separation technique used to characterize starches, but low exclusion limits and shear scission may bias results. AF4 has the potential to reduce artifacts observed in SEC such as changes in MM and size distributions as a result of shear degradation or aggregation and large branched polymers that co-elute with smaller components (4,20). A more in-depth comparison of AF4 and SEC as separation techniques for starches is available (31). To characterize size distributions and gain structural information for a commercial starch and a waxy yam starch, Perez et al. compared AF4–MALS–dRI and SEC–MALS–dRI (32). AF4 and SEC results both yielded smaller sizes for the commercial starch than the waxy yam starch, while a more quantitative recovery for AF4 (100%) was seen compared to SEC (62%). Structural characterization of the starches was also accomplished by SEC and AF4. Plotting the rg and MM of the same fraction, and using the exponent, vg, from the equation rgi = KgMivg where Kg is a constant, the polymer shape can be described (vgof 0.3, 0.5–0.6, and 1 describe the polymer shape for a sphere, a linear random coil, and a rod, respectively). Values for AF4 and SEC were all close to 0.4, which fell between a sphere and a random coil. Rolland-Sabaté et al. examined the differences between hydrodynamic chromatography (HDC)–SEC and AF4 for characterizing starches (33). Better separation of amylose and amylopectin was achieved with AF4 and allowed determination of dh and MM distributions and better structural characterization (especially for large amylopectin fractions). In addition, the branching parameter distributions showed that WTPS and WTRS amylopectins could be discerned by AF4, but not HDC–SEC.
Characterizing aggregates is important for understanding the solution behaviour and physical properties of polysaccharides. Arabinoxylan and its aggregates were characterized by AF4–MALS–dRI and SEC–MALS–dRI (34). Although aggregate concentrations were low, co-elution of individual polymers and aggregates in SEC led to larger molar masses and rg's reported compared to AF4. The MM, size, and conformation of dextrans with varying amounts of α(1-3) glycosidic linkages has also been investigated (35). Using vg values, dextrans containing the most α(1-3) linkages were found to be the smallest and densest while dextrans with the least amount displayed a quasi-linear conformation. Understanding the physical and structural properties of glucan allows for further development of biomaterials.
β-glucan's solution behaviour and ability to form aggregates may be associated with beneficial health effects. For example, understanding β-glucan digestion can aid in comprehending the physiological effects of soluble fibre. Several recent studies have used AF4–MALS–dRI to analyze β-glucans (36–38). In one of the studies, β-glucan aggregates under gastric digestion conditions were disrupted while, after undergoing small intestinal digestion, aggregates were reformed (Figure 6) (36). The disruption and re-formation of aggregates is likely to impact the behaviour and function of β-glucan. To demonstrate the effect of processing and storage on aggregates, Ulmius et al. subjected barley β-glucan samples to several conditions (such as storage time, heating, freeze time, freeze-thaw, and change in solution conditions) and performed AF4–MALS–dRI analysis (37). Disruption, structural change, or elimination of β-glucan aggregates was observed under most conditions. Properties of individual and aggregated β-glucans from oat and barley were also compared using AF4–MALS–dRI (38). Individual molecules could be distinguished from supramolecular species based on conformational differences across the size distribution. In addition, dissolution of both β-glucans under harsh alkaline conditions showed that barley β-glucan aggregates were not dissolved as previously proposed.


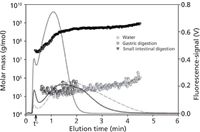
Figure 6: AF4 fractograms of barley β-glucan (lines represent fluorescence and symbols represent molar mass). Samples dispersed in water (grey-dashed line, circles), after in vitro gastric digestion (grey full line, squares), and undergoing additional small intestinal digestion (black line, triangles) were analyzed by AF4âMALS. Gastric digestion samples show a reduction in aggregate species, while the re-formation of higher density is shown after small intestinal digestion. Adapted and reproduced with permission from M. Ulmius, S. Adapa, G. Onning, and L. Nilsson,(2012), Food Chem. 130, 536â540.
AF4 has been applied to hyaluronan (HA) and sodium hyaluronate (NaHA) polysaccharides, which have important biological functions and industrial applications (39). Characterization of HA MM and conformation by AF4-MALS-dRI yielded results that were consistent with other methods, including SEC–MALS–dRI. Both AF4 and SEC were able to measure low MM (<1 × 106) samples. Molar mass distributions, an important parameter for HA characterization, were also similar between AF4 and SEC measurements. NaHA is used commercially in pharmaceutical and cosmetic products (40). Molar mass distributions and structural properties of NaHA and commercially blended NaHA mixtures were characterized and compared by frit inlet (FI) AF4–MALS–dRI. Frit inlet is a particularly gentle FFF method without the initial focusing step. Significant aggregation was not observed while samples subjected to gamma ray sterilization showed a significant breakdown of NaHA. Exudate gums are complex polysaccharides with industrial applications. They are used as emulsifiers and stabilizers and contain a small amount of proteinaceous material. Molar mass, rg, dh, conformation, apparent densities, and distribution of proteinaceous material were determined for gum arabic (GA) and mesquite gum (MG) by AF4–MALS–dRI (41). The separation of polysaccharide and proteinaceous populations and the characterization of important molecular data over the entire size range were demonstrated by AF4. Using AF4, it was possible to conclude that GA-stabilized emulsions were more stable against coalescence than MG-stabilized emulsions.
The characterization of gelatine by AF4 has also been demonstrated (42). In denatured native gelatine an increase in MM during renaturation was attributed to α-, β-, and γ-chain interactions. However, an increase in MM for thermally pre-treated gelatine was not seen, indicating an inhibition of α-, β-, and γ-chains in gelatine and therefore limiting renaturation. The effect of available lysine (lysine with a hydrogen-bonding amino group) on the formation of HMM compounds in gelatine was also characterized by AF4 (43). A decrease in available lysine with thermal treatment led to higher MMs.
Tannins play an important role in the colour, taste, and overall quality of wine. Oxidized tannins formed macromolecules and were characterized by AF4–MALS, showing soluble and insoluble populations (44). Both AF4 and small-angle X-ray scattering (SAXS) showed that the MM of insoluble macromolecules was much higher than the soluble macromolecules.
Synthetic Polymers
In recent years, advances in the characterization of synthetic polymers have included the introduction of an elevated temperature AF4 instrument and new applications in AF4 and ThFFF. Low MM polyethylene samples and a number of narrowly distributed polystyrene standards were analyzed by AF4–MALS–dRI in organic solvent and compared to SEC–MALS–dRI (45). At ambient temperature, low-density polyethylene, polypropylene, and polybutadiene containing high degrees of branching and HMMs were analyzed. As a result of the co-elution of large and small macromolecules in SEC, a correct calculation of the MM distribution and the MM average or branching ratio was not possible (Figure 7). In contrast, AF4 allows the precise determination of the MM distribution, the MM averages, and the degree of branching because the MM versus elution volume curve and the conformation plot were not affected by the co-elution issues encountered in the SEC analysis.



Figure 7: Separation of a low density polyethylene sample (CSTR-LDPE 1) by (a) HTâAF4 and (b) HTâSEC, with MALS and dRI detection. The abnormal curvature of the molar mass from HTâSEC indicates co-elution as a result of the high branching of the polymers. AF4 shows complete separation over the entire size range into the ultrahigh molar mass range not detected by SEC because of shear degradation. Adapted and reproducd with permission from T. Otte, H. Pasch, T. Macko, R. Brull, F.J. Stadler, J. Kaschta, F. Becker, and M. Buback, (2011), J. Chromatogr. A. 1218, 4257â4267. © Elsevier.
In addition, because of the absence of significant shear degradation in the channel, characterization of linear and branched HMM polyethylene by AF4 has been developed under high temperature (HT) conditions (145 °C) in organic solvent (1,2,4-trichlorobenzene [TCB]) (46). Compared to HT-SEC, HT–AF4 allows for a more complete separation of highly branched polyethylene with limited co-elution of large and small macromolecules. The HT–AF4 technique coupled with MALS detection was used for quantification and size determination of the co-eluting molecules. Furthermore, HT–AF4 induced lower shear and thermo-oxidative degradation of HMM PE and PP than HT–SEC (47). As a consequence, the HMM averages obtained from HT–AF4 are significantly higher than those obtained from HT–SEC. It was shown that most of the observed limitations of SEC could be overcome by using AF4.
AF4 has also been applied to dendritic polymer characterization. Different poly(amidoamine) (PAMAM) dendrimers have been characterized by AF4–MALS (48). The separation between different generations (4 to 9) of PAMAM particles has been shown under different pH conditions and AF4 highlighted the presence of some impurities. Coupled with other on-line characterization techniques (for example, MALS or a differential viscometer), AF4 allows for a more detailed physical characterization of each separated size fraction. Aggregation and complexation of dendritic glycopolymers used as drug delivery systems has been demonstrated using AF4–MALS (49–51). In addition, removal of small sample components through the ultrafiltration membrane during AF4 can be used to quantitatively determine the amount of complexed small guest dye molecules in core–shell polymers. This feature of AF4 can potentially be used for the separation and quantification of drugs encapsulated in polymers and makes the AF4 technique very promising for the analysis of drug delivery systems.
Other types of drug delivery systems such as micelles have been characterized by AF4. Poly(ethyleneoxide-b-ε-caprolactone) (PEO-b-PCL) self-assemblies in water were characterized by AF4 with on-line MALS–dRI–UV–vis–QELS detection (52). This study underlined the impact of the mass of the PEO and PCL fragments on the micelle size. Hydrodynamic radii measured by QELS were in good agreement with values calculated by AF4 retention times. AF4 illustrated that in some instances the number of self-assemblies present was very low compared to the number of unassembled diblock copolymers. Finally, quantification of photosensitizers used in photodynamic therapy encapsulated by these micelles has been performed. This approach was used to characterize several diblock copolymer micelles (PEG-PVP, PEG-PLA, PEG-PLGA, and PEG-PCL) and determine their in vitro half-lives in human serum (53). The impact of human serum on the micelle size and stability was shown by AF4. Indeed, micelle disassembly was observed for PEG-PVP micelles, while PEG-PLA, PEG-PLGA, and PEG-PCL micelles were far more stable.
ThFFF has been mainly used to fractionate and characterize lipophilic polymers in organic solvents. The applied force is a temperature gradient that causes thermal diffusion of analytes. The magnitude of the thermal diffusion coefficient DT has been empirically observed to depend on the polymer—solvent interface and other factors (54–56). Thermal diffusion in liquids is a complex phenomenon that is not yet fully understood (57–60). However, its usefulness in ThFFF polymer separations has been demonstrated and new interesting capabilities are being developed. For example, the observation that different polymer chemistries in the same solvent or the same polymer chemistry in different solvents can have different DT and hence tr (see Equation 2) allows for chemical composition (in addition to size) analyses of polymers.
ThFFF coupled with MALS–dRI–QELS was used to simultaneously determine the MM and composition of polystyrene–poly(n-butyl acrylate) (PS-PBA) and polystyrene-poly(methyl acrylate) (PS-PMA) copolymers (Figure 8[a] and 8[b]) (56). Equation 2 shows that the retention time is proportional to DT/D. If D can be measured independently, that is, by QELS, DT can be calculated. When on-line D measurements are made, DT can be calculated as a function of tr and subsequently correlated with polymer composition. Using this premise, the DT was found to be independent of MM for copolymers with similar compositions and dependent on composition of copolymers with similar MM in a non-selective solvent. The ThFFF–MALS–dRI–QELS combination allowed rapid determination of copolymer MM and chemical composition distributions. ThFFF has recently been coupled to NMR off-line (61) and on-line (62) in the analysis of triblock copolymers and PS, poly(methyl methacrylate) (PMMA), polyisoprene (PI), and PS-b-PMMA block copolymers, respectively. NMR provided an independent measurement of copolymer composition and confirmed compositional separation by ThFFF.



Figure 8: Weight percent composition of (a) PS-PBA and (b) PS-PMA copolymers were determined through averaged on-line DT measurements. ThFFF weight percent values are consistent with the nominal weight percent values. Adapted and reprinted with permission from J.R. Runyon and S.K.R. Williams, (2011), J. Chromatogr. A. 1218, 6774 â6779. © Elsevier.
To date, ThFFF method development has been predominantly through trial-and-error based on other published work. A recent paper demonstrated that a theoretical approach based on temperature-dependent osmotic pressure gradient and polymer–solvent interaction parameters can be used to successfully estimate DT and retention times for different polymer–solvent pairs (57). Experiments confirmed the calculation of poly(n-butyl acrylate) (PBA), poly(methyl acrylate) (PMA), and PS retention times in different solvents. This provides a potential route to predicting good solvents for polymer retention.
Thermal diffusion is an intriguing phenomenon with hidden potential for other important analyses. A recent development has shown that the correlation between theoretical and experimental DT values can provide information about the number of chain ends for branched polymers (63). The uniqueness of this study lies in the fact that the chain ends can be determined without the need for a linear polymer analogue. The ThFFF–MALS–dRI–QELS combination allows simultaneous determination of MM, composition, and number of chain ends.
Conclusions
FFF is a versatile family of techniques for characterizing biological, natural, and synthetic macromolecules. As a complementary technique to SEC, more detailed macromolecule characterizations are possible using both FFF and SEC. The open channel design and soft separation mechanism of FFF make it a powerful technique for analyzing weak macromolecule interactions, polymer aggregates, and HMM and highly branched polymers. The benefits to users are also evident in simplified sample preparations, ultrafiltration of contaminants during separation, and flexibility in carrier fluid choice among others. A conference dedicated to this subject — The 16th International Symposium on Field- and Flow-Based Separations (FFF2013) — was held in Pau, France in July, and FFF2014 will be held in Salt Lake City, Utah, USA in October next year. Further interesting developments are anticipated, along with a flurry of associated publications.
Acknowledgements
CB and SKRW thank the National Science Foundation CHE-1013029 for financial support.
Carmen Bria is a PhD student in the Chemistry Department at the Colorado School of Mines (Colorado, USA). His research focuses on the use of FFF and light scattering to characterize proteins and probe protein aggregation processes. He is also working on developing improved membrane surfaces for flow FFF.
Frédéric Violleau graduated from ENSCT (INPT – University of Toulouse, France) with a "Diplôme d'Ingénieur en chimie" (equivalent to a MSc in chemistry) and from the National Polytechnic Institute of Toulouse (University of Toulouse) with a PhD in organic chemistry. He joined Ecole d'Ingénieurs de PURPAN (EI Purpan – INPT – University of Toulouse) in 2003 and he is currently vice head of the Agricultural and Food Sciences Department. He has experience in using AsFlFFF technology for various applications involving proteins, polysaccharides, polymers, and particles.
S. Kim R. Williams is a professor of chemistry and the Director of the Laboratory for Advanced Separations Technologies at the Colorado School of Mines. She began her journey with FFF as a postdoctoral fellow with the late J. Calvin Giddings at the University of Utah (Utah, USA) and has acquired more than 25 years of experience in this field. Research in the Williams group focuses on developing new capabilities for nanoparticle and macromolecular analyses using FFF and related methods. Dissemination of these new technologies are done through collaborations with scientists at universities, companies, and national laboratories. She recently edited a book entitled Field-Flow Fractionation in Biopolymer Analysis.8
References
1. T. Arakawa, D. Ejima, T.S. Li, and J.S. Phil, J. Pharm. Sci. 99(4), 1674–1692 (2010).
2. B. Demeule, M.J. Lawrence, A.F. Drake, R. Gurny, and T. Arvinte, Biochim. Biophys. Acta-Proteins. Proteomics 1774(1), 146–153 (2007).
3. A. Hawe, S. Romeijn, V. Filipe, and W. Jiskoot, J. Pharm. Sci. 101(11), 4129–4139 (2012).
4. S. Podzimek, P. Lebedat, and C. Johann, LCGC North America 27(1), 62–69 (2009).
5. D. Lee and S.K.R. Williams, J. Chromatogr. A. 1217(10), 1667–1673 (2010).
6. R.N. Qureshi and W.T. Kok, Anal. Bioanal. Chem. 399(4), 1401–1411 (2011).
7. G. Yohannes, M. Jussila, K. Hartonen, and M.L. Riekkola, J. Chromatogr. A. 1218(27), 4104–4116 (2011).
8. S.K.R. Williams and K.D. Caldwell, Eds., Field-Flow Fractionation in Biopolymer Analysis (Springer Verlag, Wien, 2012).
9. J. Pollastrini, T.M. Dillon, P. Bondarenko, and R.Y.T. Chou, Anal. Biochem. 414(1), 88–98 (2011).
10. J.Y. Lee, D. Choi, C. Johan, and M.H. Moon, J. Chromatogr. A. 1218(27), 4144–4148 (2011).
11. K. Rebolj, D. Pahovnik, and E. Zagar, Anal. Chem. 84(17), 7374–83 (2012).
12. J.Y. Kim, S.-K. Kim, D. Kang, and M.H. Moon, Anal. Chem. 84(12), 5343–5350 (2012).
13. R.N. Qureshi, E. Kaal, H.G. Janssen, P.J. Schoenmakers, and W.T. Kok, Anal. Chim. Acta 706(2), 361–366 (2011).
14. K.H. Kim, J.Y. Lee, and M.H. Moon, Analyst 136(2), 388–392 (2011).
15. K.H. Kim, J.Y. Kim, M.O. Kim, and M.H. Moon, J. Proteomics. 75(8), 2297–2305 (2012).
16. K.H. Kim and M.H. Moon, Anal. Chem. 83(22), 8652–8658 (2011).
17. M. Hassellov, G. Hulthe, B. Lyven, and G. Stenhagen, J. Liq. Chromatogr. Relat. Technol. 20(16–17), 2843–2856 (1997).
18. P. Reschiglian, A. Zattoni, B. Roda, L. Cinque, D. Parisi, A. Roda, F. Dal Piaz, M.H. Moon, and B.R. Min, Anal. Chem. 77(1), 47–56 (2005).
19. K.H. Kim, J.Y. Lee, S. Lim, and M.H. Moon, J. Chromatogr. A. 1280(0), 92–97 (2013).
20. M.J. Gidley, I. Hanashiro, N.M. Hani, S.E. Hill, A. Huber, J.-L. Jane, Q. Liu, G.A. Morris, A. Rolland-Sabaté, A.M. Striegel, and R.G. Gilbert, Carbohydr. Polym. 79(2), 255–261 (2010).
21. S. You, S.G. Stevenson, M.S. Izydorczyk, and K.R. Preston, Cereal. Chem. 79(5), 624–630 (2002).
22. M. Van Bruijnsvoort, K.G. Wahlund, G. Nilsson, and W.T. Kok, J. Chromatogr. A. 925(1), 171–182 (2001).
23. L. Nilsson, Food Hydrocolloids 30(1), 1–11 (2013).
24. K.G. Wahlund, M. Leeman, and S. Santacruz, Anal. Bioanal. Chem. 399(4), 1455–1465 (2011).
25. S. Juna, P.A. Williams, and S. Davies, Carbohydr. Polym. 83(3), 1384–1396 (2011).
26. S. Juna and A. Huber, Starch-Starke 64(1), 18–26 (2012).
27. S. Juna and A. Huber, Starch-Starke 64(2), 87–96 (2012).
28. S. Juna and A. Huber, J. Chromatogr. A. 1219, 161–172 (2012).
29. S. Juna and A. Huber, Starch-Starke 64(3), 171 (2012).
30. S. Juna, M. Damm, C.O. Kappe, and A. Huber, Starch-Starke 64(8), (2012).
31. H. Ruebsam, M. Krottenthaler, M. Gastl, and T. Becker, Starch-Starke 64(9), 683–695 (2012).
32. E. Perez, O. Gibert, A. Rolland-Sabate, Y. Jimenez, T. Sanchez, A. Giraldo, B. Pontoire, S. Guilois, M.C. Lahon, M. Reynes, and D. Dufour, J. Agric. Food Chem. 59(1), 263–273 (2011).
33. A. Rolland-Sabate, S. Guilois, B. Jaillais, and P. Colonna, Anal. Bioanal. Chem. 399(4), 1493–1505 (2011).
34. L. Pitkanen, M. Tenkanen, and P. Tuomainen, Anal. Bioanal. Chem. 399(4), 1467–1472 (2011).
35. R. Irague, A. Rolland-Sabate, L. Tarquis, J.L. Doublier, C. Moulis, P. Monsan, M. Remaud-Simeon, G. Potocki-Veronese, and A. Buleon, Biomacromolecules 13(1), 187–195 (2012).
36. M. Ulmius, S. Adapa, G. Onning, and L. Nilsson, Food Chem. 130(3), 536–540 (2012).
37. M. Ulmius, G. Onning, and L. Nilsson, Food Hydrocolloids 26(1), 175–180 (2012).
38. A. Hakansson, M. Ulmius, and L. Nilsson, Carbohydr. Polym. 87(1), 518–523 (2012).
39. T. Luan, Y.P. Fang, S. Al-Assaf, G.O. Phillips, and H.B. Zhang, Polymer. (Guildf). 52(24), 5648–5658 (2011).
40. M. Ali, E. Hwang, I.H. Cho, and M.H. Moon, Anal. Bioanal. Chem. 402(3), 1269–1276 (2012).
41. J. Alftren, J.M. Penarrieta, B. Bergenstahl, and L. Nilsson, Food Hydrocolloids 26(1), 54–62 (2012).
42. K. Rbii, O. Surel, N. Brambati, A.M. Buchert, and F. Violleau, Food Hydrocolloids 25(3), 511–514 (2011).
43. K. Rbii, F. Violleau, N. Brambati, A.M. Buchert, and O. Surel, Food Hydrocolloids 25(6), 1409–1412 (2011).
44. A. Vernhet, S. Dubascoux, B. Cabane, H. Fulcrand, E. Dubreucq, and C. Poncet-Legrand, Anal. Bioanal. Chem. 401(5), 1559–1569 (2011).
45. T. Otte, H. Pasch, T. Macko, R. Brull, F.J. Stadler, J. Kaschta, F. Becker, and M. Buback, J. Chromatogr. A. 1218(27), 4257–4267 (2011).
46. T. Otte, T. Klein, R. Brull, T. Macko, and H. Pasch, J. Chromatogr. A. 1218(27), 4240–4248 (2011).
47. T. Otte, H. Pasch, R. Brull, and T. Macko, Macromol. Chem. Phys. 212(4), 401–410 (2011).
48. I.J. Suarez, R. Rosal, A. Rodriguez, A. Ucles, A.R. Fernandez-Alba, M.D. Hernando, and E. Garcia-Calvo, TrAC Trends. Anal. Chem. 30(3), 492–506 (2011).
49. A. Lederer and S. Boye, LCGC Europe 24(12), 620–628 (2011).
50. S. Boye, N. Polikarpov, D. Appelhans, and A. Lederer, J. Chromatogr. A. 1217(29), 4841–4849 (2010).
51. S. Boye, D. Appelhans, V. Boyko, S. Zschoche, H. Komber, P. Friedel, P. Formanek, A. Janke, B.I. Voit, and A. Lederer, Biomacromolecules 13(12), 4222–4235 (2012).
52. J. Ehrhart, A.F. Mingotaud, and F. Violleau, J. Chromatogr. A. 1218(27), 4249–4256 (2011).
53. T. Miller, R. Rachel, A. Besheer, S. Uezguen, M. Weigandt, and A. Goepferich, Pharm. Res. 29(2), 448–459 (2012).
54. R.M. Sisson and J.C. Giddings, Anal. Chem. 66(22), 4043–4053 (1994).
55. M.E. Schimpf and J.C. Giddings, J. Polym. Sci Part. B: Polym. Phys. 27(6), 1317–1332 (1989).
56. J.R. Runyon and S.K.R. Williams, J. Chromatogr. A. 1218(38), 6774–6779 (2011).
57. J.R. Runyon and S.K.R. Williams, J. Chromatogr. A. 1218(39), 7016–7022 (2011).
58. M.E. Schimpf and S.N. Semenov, J. Phys. Chem. B. 104(42), 9935–9942 (2000).
59. E.P.C. Mes, W.T. Kok, and R. Tijssen, Int. J. Polym. Anal. Charact. 8(2), 133–153 (2003).
60. A.C. Wurger, Phys. Rev. Lett. 102(7), (2009).
61. C.A. Ponyik, D.T. Wu, and S.K.R. Williams, Anal. Bioanal. Chem. In Press (2013).
62. W. Hiller, W. van Aswegen, M. Hehn, and H. Pasch, Macromolecules. 46(7), 2544–2552 (2013).
63. J.R. Runyon, D.T. Wu, and S.K.R. Williams, Characterizing branched Polymers Using Thermal Field-Flow Fractionation — In Preparation.





.")



USP CEO Discusses Quality and Partnership in Pharma
December 11th 2024Ronald Piervincenzi, chief executive officer of the United States Pharmacoepia, focused on how collaboration and component quality can improve worldwide pharmaceutical production standards during a lecture at the Eastern Analytical Symposium (EAS) last month.


The LCGC Blog: Historical (Analytical) Chemistry Landmarks
November 1st 2024The American Chemical Society’s National Historic Chemical Landmarks program highlights sites and people that are important to the field of chemistry. How are analytical chemistry and separation science recognized within this program?

Leveraging an Enterprise Laboratory Informatics Platform to Maximize Scientific Data Advantage
September 9th 2024As data volumes and expectations for fast scientific discovery continue to increase, laboratory-based research organizations can no longer rely on a siloed approach to data management. To remain competitive, scientific organizations need to connect all their data, from discovery through manufacturing, in a unified informatics platform.

