Drawing A Better Map: Recent Advances in Protein Digestion and Peptide Mapping
LCGC Europe
Biotherapeutics have become the hottest topic in pharmaceutical research over the past decade. With the increased interest in biotherapeutics, there has been a concomitant increase in new analytical methods for characterizing these large, complex molecules. This installment of “Column Watch” discusses advances in “bottom-up” analysis of monoclonal antibodies, while highlighting the role and importance column chemistry still plays in developing highly selective high-performance liquid chromatography (HPLC) methods for peptides.
Biotherapeutics have become the hottest topic in pharmaceutical research over the past decade. With the increased interest in biotherapeutics, there has been a concomitant increase in new analytical methods for characterizing these large, complex molecules. This installment of “Column Watch” discusses advances in “bottom-up” analysis of monoclonal antibodies, while highlighting the role and importance column chemistry still plays in developing highly selective high-performance liquid chromatography (HPLC) methods for peptides.
Monoclonal antibodies (mAbs) are a type of biotherapeutic that is seeing a high degree of interest from academia and industry. Since the first approval in 1986, nearly 60 antibodies have been manufactured and commercialized up until 2015. 2016 alone was a banner year for antibody-based therapeutics, as the FDA approved seven therapeutic antibodies as new molecular entities: monoclonal antibodies are used to fight a plethora of diseases ranging from inflammatory conditions (1), autoimmune diseases (2), various types of cancers (3), and other infectious and neurological diseases (4).
Monoclonal antibodies are complex molecular entities with molecular weights of approximately 150 kDa. These molecules are composed of amino acids that are connected through amide (peptide) bonds. Monoclonal antibodies are expressed in cell culture systems, meaning that these molecules are susceptible to an entire suite of post-translational modifications (PTMs) of an enzymatic or non-enzymatic nature. The most common PTMs to a mAb include N-terminal pyroglutamate formation, methionine oxidation, asparagine deamidation, aspartic acid isomerization, C-terminal lysine truncation, and glycosylation (5). As a result of the susceptibility of mAbs to be modified, the resultant product, directly from the expression system, is often a heterogeneous mixture of variants of the mAb. This high degree of complexity of the resultant product necessitates the need for analytical methods for characterizing mAbs.
One of the most useful techniques for characterizing post-translational modifications is to perform “bottom-up” analyses of proteins or mAbs, also called peptide mapping. In peptide mapping, the mAb is digested by a protease, like trypsin, into peptides. These peptides are subsequently separated and identified by liquid chromatography–mass spectrometry (LC–MS). However, because of the complex folding and disulfide bonding present with mAbs, before digestion occurs, these proteins are typically denatured, or unfolded, to allow full access of the protease to the consensus sequence on the protein. In addition, disulfide bonds are reduced and alkylated to prevent spontaneous refolding of the protein. Once these steps are completed, the protease is added to the sample, and the digestion reaction can proceed from anywhere between four to twenty-four hours. After this pre-determined amount of time, the reaction is quenched by addition of formic acid, and the resultant sample is diluted and prepared for reversed-phase LC analysis.
The process of digesting a protein, though, can lend itself to artifacts in the peptide mapping results. Usually, this is observed through additional methionine oxidation and asparagine deamidation that is not due to the inherent cellular machinery used to express the mAb. Heating of the digested sample over an extended period, over digestion by the protease, and reactive intermediates generated during the digestion reacting with peptides may cause artefactual modifications of the peptides (6).
Peptide maps of proteins can have anywhere from tens to hundreds of peaks. Therefore, chromatographic methods with high resolving ability or peak capacity are a necessity. One way to calculate resolution is through Equation 1, where Rs is resolution, âtR is differential migration, and WA and WB are the peak widths of analyte A and B, respectively.
Rs = âtR/0.5(WA + WB) [1]
Resolution of peaks in a chromatographic separation comes from two factors: a thermodynamic (chemical) factor, and a kinetic (physical) factor. The kinetic factor (the denominator of Equation 1) is due mostly to the particle architecture and how well the column is packed. For proteins, the advent of superficially porous particles (SPPs) has drastically increased the efficiency of methods that were originally developed on fully porous particles (FPPs). For more information on why such advantages are present with SPP-packed columns, the interested reader may consult the following review articles (7, 8).
The thermodynamic factor that mostly affects resolution is the type of ligand that is used in reversed-phase liquid chromatography. Chromatographic generation of a peptide map has typically been performed on a C18 column. Just like with small molecule chromatography, the types of molecular interactions that a chromatographer can take advantage of with a C18 column are mostly dispersive interactions and, to a lesser controlled extent, ionic interactions from silanols on the base silica (9). However, peptides are amphipathic molecules, having both a hydrophobic and hydrophilic domain. It is hypothesized that the hydrophilic domains of peptides could interact with more polar reversed-phase columns through dipole-dipole interactions or hydrogen bonding. With more interactions at the chromatographer’s disposal, more selective methods could be developed that permit better retention of polar peptides or orthogonal selectivity to conventional C18 chromatography. For the rest of this installment of “Column Watch,” experiments will be described with the goal of obtaining better peptide maps. First, different digestion protocols are assessed to optimize tryptic digest conditions. Then, the role of phase chemistry will be examined to better resolve critical pairs of peptides that may be present in a digested protein sample.
Experimental Procedure
Tryptic Digest with SOLu-Trypsin
Since SigmaMAb is a glycosylated immunoglobulin G1 (IgG1) antibody, the antibody needed to be deglycosylated prior to digestion. SigmaMAb was resuspended by adding 1 mL of “reaction buffer” (10 mM sodium phosphate, 150 mM sodium chloride, pH 7.4). Glycinator (Genovis), an endoglycosidase, was resuspended by adding 50 µL of LC–MS grade water. Fifty microliters of Glycinator was mixed with SigmaMAb. The mAb-endoglycosidase mixture was heated at 37 °C for 35 min. Afterwards, using a HisTrap spin column, Glycinator was removed from the deglycosylated mAb.
Prior to digestion, the deglycosylated mAb was dried by vacuum centrifugation. A denaturing solution consisting of a 1:1 mixture of 40% trifluoroethanol and 20 mM dithiothreitol (DTT) in 50 mM ammonium bicarbonate was prepared, and 20 µL of this solution was added to the dried mAb. The mAb plus denaturant mixture was incubated at 57 °C for one hour. Upon completion of the denaturation step, the reduced mAb was alkylated by mixing 5.0 µL of 200 mM iodoacetamide in 50 mM ammonium bicarbonate. The alkylation reaction occurred for one hour in the dark at room temperature. Upon completion of the alkylation reaction, 220 µL of 50 mM ammonium bicarbonate was added to dilute the denaturants. SOLu-Trypsin was added to the reduced and alkylated mAb such that the ratio of trypsin–mAb was 1:20 by mass. The trypsin-mAb solution was incubated at 37 °C at various time points (Figure 1). After each time point, the digestion reaction was quenched by adding 2.0 µL of formic acid, the sample was centrifuged at 5,000 x g for 30 s, and the supernatant was collected. Prior to analysis, the sample was diluted 1:1 with water containing 0.1% (v/v) difluoroacetic acid (DFA) prior to LC–MS analysis.


Tryptic Digest with SOLu-Trypsin and Rapid Digest Buffer
Deglycosylation, reduction, and alkylation was performed using the same procedure as in the previous two paragraphs. To the denatured mAb, 500 µL of Rapid Digest buffer was added to the sample. SOLu-Trypsin was added such that the ratio of trypsin: mAb was 1:20. After addition of SOLu-trypsin, the trypsin-mAb sample was incubated at 60 °C for one hour. After the one-hour digest, the reaction was quenched by mixing 2.0 µL of formic acid to the sample, centrifuging the sample at 5,000 x g for 30 s, and collecting the supernatant. The sample was then diluted 1:1 with water containing 0.1% (v/v) difluoroacetic acid prior to LC–MS analysis.
Peptide Selectivity Study (“Kappa-Kappa Study”)
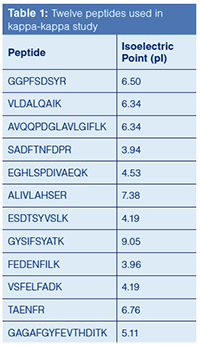
A standard peptide mix, containing 14 peptides, was assayed using the HPLC conditions listed in Figure 4. Table 1 lists the identity of the 12 peptides used in the study (two of the peptides in the 14-component mix were not observed in the study).
Results
Rapid Digestion for High-Throughput Peptide Mapping
Since one of the ways that artifacts can present themselves during a peptide map experiment is through over-digestion of the protein, steps were taken to decrease the digestion time as much as possible. At first, the use of a recombinant trypsin was used, over a conventional, animal derived trypsin. Figure 1 displays the results of a time course study where aliquots of the digested mAb were taken, the aliquot was assayed by RPC, and the number of peptide peaks were determined. Note that with the recombinant trypsin, only four hours were required to attain complete digestion of the mAb whereas the standard trypsin required 18 hours.

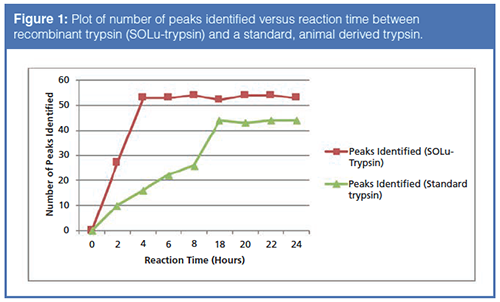

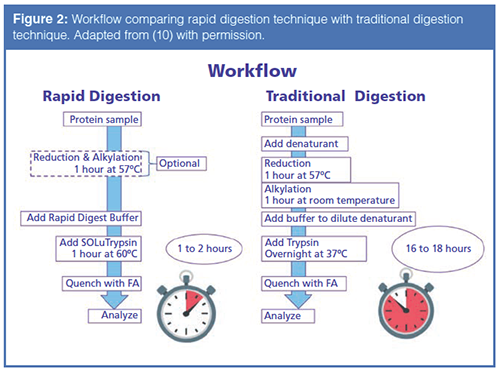
Even though the reaction time has dropped to four hours, oxidation may still occur on some of the peptide fragments due to the fast reaction kinetics of oxidizing surface amino acids like methionine. Oxidation caused by the digestion protocol could, in a biopharmaceutical setting, cause the rejection of a qualified lot of therapeutic drug. In recent years, some vendors have developed “Rapid Digest Kits” for performing peptide mapping experiments, with an eye for high-throughput. Therefore, a recombinant trypsin coupled with a “Rapid Digest Kit” was employed, allowing for the complete digestion and analysis of the mAb in approximately two hours with only a one-hour digestion time. Figure 2 displays a comparison of the workflow of the “rapid” method versus conventional methods (10). Again, note the drastic difference in time between the two techniques.

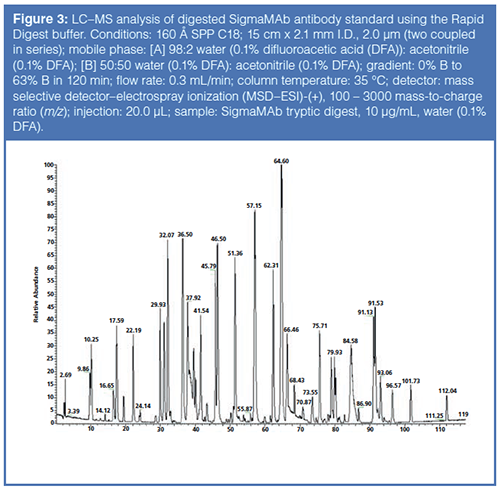
Upon completion of the Rapid Digestion technique, the digested sample was subjected to LC–MS analysis. Figure 3 displays the results of the analysis. After data analysis of the sample prepared with the Rapid Digest buffer, the sequence coverage of the heavy and light chain was 99% and 97% respectively. Standard digestion protocols, without the Rapid Digest buffer, yielded sequence coverage of the heavy and light chains of 88% and 94%, respectively.
Peptide Selectivity – Do We Need More than C18?
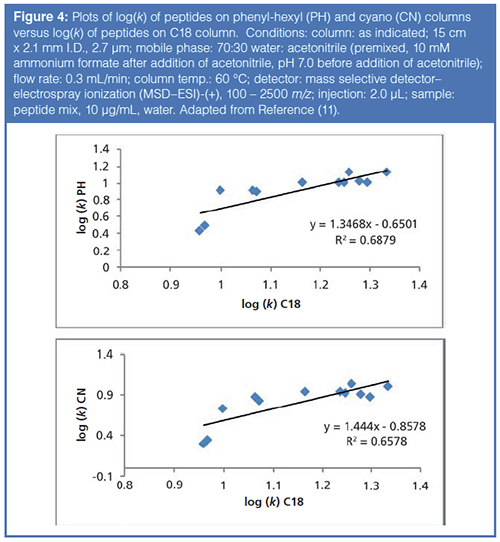
As noted earlier in the article, most peptide mapping experiments use C18 columns for the LC separation. However, peptides have many polar functional groups that, theoretically, could interact with more polar ligands (i.e. RP-Amide, F5, Phenyl-hexyl, etc.). Previous work (11) has hinted at the possibility for orthogonal selectivity of peptides on different phase chemistries to conventional C18 phase chemistries. Figure 4 illustrates this concept with plots of the logarithm of the retention factors for a set of peptide probes assayed on a C18 column versus a phenyl-hexyl (PH) and a cyano (CN) column (so-called kappa-kappa plots). From examination of Figure 4, one can see that there are differences in retention on the different phase chemistries. The phenyl-hexyl column, with π – π interactions and shape selectivity from the rigid, aromatic ring, brings in additional molecular interactions not present on a C18 column. In addition, the dipole-dipole interactions and π – π interactions of the CN phase also contribute to selectivity that is orthogonal to a C18 column. With peptides being composed of amino acids with varied side chains, these polar interactions may contribute to differential selectivity.

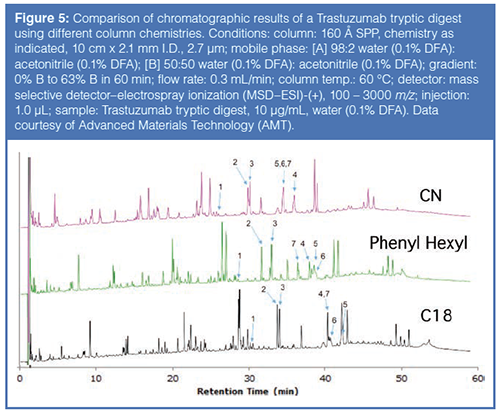
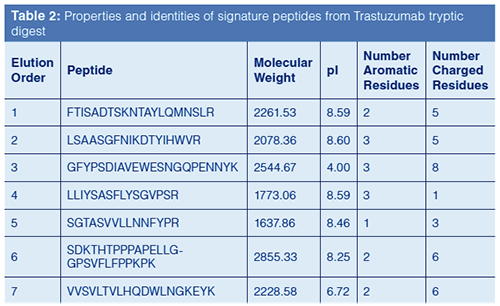
Based on this previous work, columns with different phase chemistries were used to improve the resolution of peptides resulting from a tryptic digest using the Rapid Digest technique. Figure 5 compares chromatograms generated from a tryptic digest of Trastuzumab, a monoclonal antibody used to treat various head, neck, and breast cancers. Table 2 provides the identity of the “signature” peptides that are numbered in Figure 5. As can be noted from the chromatographic data, each column yielded a different elution profile for the signature peptides. In fact, only the phenyl-hexyl column could resolve all seven signature peptides; the other two columns resulted in co-elutions of two or more peptides. These results, again, imply the advantage of using additional column chemistries compared to just C18.


Conclusion
Peptide mapping is an analytical technique where the primary structure of a protein is assessed through the digestion of the protein. Recent strategies have been developed, using faster enzymes and new buffer systems, that enable the analyst to perform peptide mapping experiments and the resultant LC–MS analysis in an afternoon whereas older techniques would take at least one to two days. These older techniques were also prone to under or over digestion as well as oxidation and deamidation due to the digestion protocol. Limiting digestion time to one to two hours minimizes these artifacts without compromising sequence coverage.
SSP-packed HPLC columns have allowed for an increase in efficiency and throughput of methods devoted to the analysis of proteins and peptides. In peptide mapping, a C18 column has mostly been used for such analyses. Using different phase chemistries can allow for orthogonal separations of peptide peaks to that obtained with a C18 column. Future research will involve screening different mobile phase systems and examining how the mobile phase can further improve resolution of peptide peaks and improve the sensitivity of methods.
References
- J. Zhen, J. Kim, Y. Zhou, E. Gaidamauskas, S. Subramanian, and P. Feng. MAbs10(7), 951–959 (2018).
- H.M. Shepard, G.L. Philips, C.D. Thanos, and M. Feldmann, Clin. Med. (Lond.) 17, 220–232 (2017).
- A. Hey, Microbiol. Spectrum3, AID–0026–2014 (2015).
- J.C. Novak, A.E. Lovett-Racke, and M.K. Racke, Arch. Neurol.65, 1162–1165 (2008).
- H. Liu, G. Ponniah, H. Zhang, C. Nowak, A. Neill, N. Gonzalez-Lopez, R. Patel, G. Cheng, A.Z. Kita, and B. Andrien. MAbs6(5), 1145–1154 (2014).
- A. Nigam, M. Subramanian, and P.K. Rajanna, Chromatographia81(1), 57–64 (2018).
- V. Gonzalez-Ruiz, A.I. Olives, and M.A. Martin, TrAC64, 17–28 (2015).
- J.J. Kirkland, S.A. Schuster, W.L. Johnson, and B.E. Boyes. J. Pharma. Anal.3, 303–312 (2013).
- L.R. Snyder, J.J. Kirkland, and J.W. Dolan, Introduction to Modern Liquid Chromatography (John Wiley and Sons, Hoboken, New Jersey, 3rd Ed., 2010).
- Z, Cao. Development of A Simple and Rapid Digestion Protocol for Proteomics Sample Preparation. Poster presentation at the American Society of Mass Spectrometry (ASMS), 2018.
- C.E. Muraco, G. Oden, and D.S. Bell. Alternate Selectivity in Biomacromolecule Separations: What Can We Learn from Small Molecule Research? Oral presentation given at the Pittsburgh Conference on Analytical Chemistry and Spectroscopy (Pittcon), 2018.
Cory E. Muraco is a Senior R&D Scientist in the Liquid Separations R&D group at MilliporeSigma, in Bellefonte, Pennsylvania. Cory completed his graduate studies at Youngstown State University in 2013, focusing on the analysis and characterization of oxidized proteins. Upon graduation, Cory joined MilliporeSigma, first joining the chemical standards R&D group, then transferring to the liquid separations R&D group. Cory’s current role at MilliporeSigma is to research, develop, and present on new particle technology for improved chromatographic separations of both small and large molecules and to develop new methodologies for characterizing biomacromolecules by several modes of chromatography. Cory has written about his research in several trade magazines and presented oral and poster presentations on his research at numerous conferences.
David S. Bell is a director of Research and Development at Restek. He also serves on the Editorial Advisory Board for LCGC and is the Editor for “Column Watch.” Over the past 20 years, he has worked directly in the chromatography industry, focusing his efforts on the design, development, and application of chromatographic stationary phases to advance gas chromatography, liquid chromatography, and related hyphenated techniques. His main objectives have been to create and promote novel separation technologies and to conduct research on molecular interactions that contribute to retention and selectivity in an array of chromatographic processes. His research results have been presented in symposia worldwide, and have resulted in numerous peer-reviewed journal and trade magazine articles. Direct correspondence to: LCGCedit@ubm.com
















Evaluating Body Odor Sampling Phases Prior to Analysis
April 23rd 2025Researchers leveraged the advantages of thermodesorption, followed by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry (GC×GC/TOF-MS), to compare and assess a variety of sampling phases for body odor.



