Developments in HPLC/UHPLC Column Technology
A discussion of new column packing materials– including sub-2-?m particles, superficially porous particles and second-generation monoliths– and their impact on instrumentation and laboratory productivity.
The last two years have seen a continuous introduction of new high performance liquid chromatography (HPLC) and ultrahigh-pressure liquid chromatography (UHPLC) packing materials, including a range of sub-2-µm particles, superficially porous particles and second-generation monoliths. Here, we look at these introductions and their impact on instrumentation and laboratory productivity. In addition, we discuss future directions in column technology.
Every two years, we publish a special supplement on high performance liquid chromatography (HPLC) column technology in LCGC North America that brings readers up to date on the latest improvements in this important area. This is the first time this supplement has been published in LCGC Europe and we have a total of four articles (in addition to this one), which discuss hybrid packings, superficially porous (core–shell) packings and hydrophilic interaction liquid chromatography (HILIC) (1–6). The last time I gave a general overview on columns was in the 2008 supplement (7). In the 2010 supplement (8), I focused on improvements and phases for reversed-phase chromatography, the most frequently used mode. This article will focus on an overview of column developments and discuss future directions in this area. Since the 2008 article, there has been a continuing interest in column developments with improvements in small porous particle technology, monoliths (both polymer- and silica-based), superficially porous particles (SPP) and a tremendous focus on HILIC. For handling complex samples, multidimensional and comprehensive LC×LC are still getting attention. In this update on column technology, I will cover some of the packing material and column developments and highlight observations made in the past four years. Other articles in this special supplement will expand on changes that have occurred in stationary phases in the various modes.


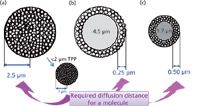
Figure 1: Comparison of diffusion distances for (a) 5-µm totally porous particles (TPP), with a sub-2-µm particle below the arrow, (b) 5-µm superficially porous particles (SPP) and (c) 2.7-µm SPP.
High-Throughput and Productivity: Still a Driving Force in Liquid-Phase Separations
Sub-2-µm Columns: One of the driving forces for continued improvements in HPLC column technology is the requirement for higher throughput without sacrificing analytical performance. One of the trends noted in the last several years is the strong interest in smaller totally porous particles (TPP), especially those with average particle diameters less than 2 µm [Figure 1(a)]. In 2012, at least 25 companies are now promoting the virtues of sub-2-µm columns (Table 1). New companies are being added each year. It is almost expected now for the introduction of new columns to provide at least one sub-2 µm particle size in the portfolio.


Table 1: Commercial 2- and sub-2-µm totally porous HPLC columns.*
When sub-2-µm porous particles are packed into short columns, separations can be performed faster, sometimes in just a minute or two, than longer columns packed with larger particles (~5 µm) without sacrificing chromatographic resolution. The flat van Deemter curves noted for sub2µm columns allow (and perhaps demand) relatively high flow rates to be used, in the range of 1.5–2 mL/min, if necessary. Even though the column pressure increases with the inverse square of the average particle diameter, these shorter columns (usually 50 mm and under) can be used with most conventional HPLC pumping systems, even at these increased flow rates.
For more demanding separations, longer columns of 100- and 150-mm lengths may be required. With such lengths, column back pressures may increase beyond the capabilities of conventional pumping systems (~400 bar upper limit). Thus, in recent years, pumping systems capable of operation at pressures as high as 20000 psi (1330 bar) have come onto the market. The advent of the term ultrahigh-pressure liquid chromatography (UHPLC) has somewhat mesmerized the chromatography world into thinking that an entirely new technology has arisen. However, it is rare to see a separation run at such high pressures, even at 1000 bar. User concerns about stress on instrument hardware and on the columns themselves have limited widespread applications at these high pressures. The demands of sample cleanliness and easier "pluggability" of the small porosity frits terminating the ends of sub-2-µm columns also have made some users cautious about jumping into their routine application. Nevertheless, these columns have proven to be rugged, even at extremely high pressures, if used properly by ensuring particle-free samples are injected. With the introductions of zero- or low-dead-volume, high-pressure guard columns and zero-dead-volume in-line filters to help protect the analytical column, liquid chromatographers are becoming more comfortable using these columns and many publications are now seen employing sub-2-µm columns, even in routine environments.
What the advent of UHPLC has done is make instrument manufacturers more aware of the need to provide systems with minimal extra column effects (that is, band broadening outside of the column itself). These small-particle columns are so efficient (~250000 plates/m) that any unswept and unnecessary volumes and connections in the flow path have to be minimized. In addition to the high-pressure capability required for the pump and other hardware components (such as injector valves, mixers, and fittings), attention also is paid to the dwell volume now (also referred to as the gradient delay volume — the volume from the point of mixing of the solvents to the head of the column). When attempting to develop 1–2 min separations, liquid chromatographers can no longer wait for a gradient that takes several minutes to reach the column because ballistic gradients are now the norm.
Some column and instrument suppliers have held off on joining the sub-2-µm bandwagon and have made more moderate reductions in the particle size from the popular 3–3.5 µm columns. The particle diameter is larger for packed columns in the range of 2–3 µm than the sub-2-µm particles, so the pressure drop is lower but efficiency is better than the more popular 3–3.5 µm particles. The arguments for using packings in the 2–3 µm range revolve around considering the entire separation cycle time: Higher temperatures improve efficiency and reduce back pressure, improved LC system hydraulics decrease band dispersion and gradient re-equilibration time, and faster autosamplers, detectors and data systems increase overall system efficiency. The use of lower pressures also places less stress on the instrumentation and column materials.
Interestingly, I have not seen even smaller totally porous particle columns coming to the market in the last four years. Currently, the smallest totally porous particle size that is commercially available is 1.5 µm and this diameter has been available for many years. However, there are other competing technologies now on the market (and coming onto the market) that do not expose the column and instrumentation to such dramatic pressure conditions: SPP columns and monoliths.
Superficially Porous (Core–Shell) Particle Packed Columns: The concept of SPP has been around since the beginning of HPLC in the late 1960s; 40–60 µm pellicular packings helped set the stage for further HPLC column development. Basically, this type of particle, also referred to as a core–shell and fused-core particle nowadays, has a solid inner core and a porous outer layer. The initial modern 5-µm SPP [Figure 1(b)], introduced in 1991, was designed for fast separations of macromolecules like proteins that, because of their low diffusion coefficients, gave poor column efficiencies with wide-pore, totally porous particle columns run at high flow rates (9). For proteins, the wide-pore SPP with a thin porous layer gave faster mass transfer into and out of the solid-core packing and thus showed improved efficiency at high flow rates.
Compared to a TPP of the same diameter, when separating small molecules with modern SPP columns [Figure 1(c)] there are a number of improvements in the overall efficiency, all related to elements of the van Deemter equation. Professor van Deemter was a Dutch scientist who came up with an equation that shows the impact of various experimental parameters on column efficiency, H or height equivalent to a theoretical plate (HETP). The lower the HETP value, the better the efficiency (that is, the more theoretical plates, N). The simplest form of the van Deemter equation (equation 1) is made up of three elements: First, the "A term," the eddy diffusion (and flow distribution) term that defines the path that the mobile phase (and consequently the solute) flows through a packed bed. Having a homogeneous packed bed without erratic flow paths minimizes this term. The spherical particles, narrow particle size distribution and proper packing techniques give rise to very homogeneous beds for the SPP. For SPP, the lower A term contributes significantly to the decrease in HETP relative to a totally porous packing of the same particle diameter. Totally porous particles generally display about 25% wider particle size distribution compared to SPP.
H(HETP) = A + B/u + C u [1]
Next, the "B term," longitudinal diffusion, has to do with solute diffusion in an axial direction. If one were to inject a solute into the middle of a column under stopped flow conditions (u [mobile phase velocity] = 0) and go off to lunch, while gone the solute would diffuse in all directions. When the chemist returned, this diffusion process would further distribute the solute in a longitudinal direction and therefore broaden the band giving rise to a higher value of HETP. The morphology of the SPP particle apparently limits this rate of diffusion and thus slightly decreases the degree of band spreading compared to a totally porous particle. The "C term" refers to solute mass transfer into and out of the particle itself; it turns out that for SPP and small molecules, this C term is only very slightly favourable compared to a totally porous particle. Overall, the combination of these three terms results in column efficiency of a 2.6–2.7 µm SPP that is not unlike that of the sub-2-µm particles. For a detailed theoretical and practical discussion of comparisons of different particle geometries, readers should refer to the excellent recently published paper by Gritti and Guiochon (10).
Probably the biggest single advantage of the modern SPP column is the lower pressure drop compared to the sub-2-µm particle packed columns. Because the most popular SPPs are in the 2.5–2.7 µm range, the pressure drop of an equal size column run under equivalent conditions is around one-half compared to a 1.7–1.8 µm column. So, the bottom line is the same column efficiency at half the operating pressure. Thus, the SPP columns are becoming quite popular as evidenced by the larger number of vendors (see Table 2) since the 2008 columns review (7). Although the number of stationary phases of the SPPs falls short of the number available for sub-2-µm particles, manufacturers are quickly adding new phases to their portfolios each year. For reversedphase separations, most applications are covered with existing C8 and C18 SPP phases.


Table 2: Superficially porous particle columns for reversed-phase chromatography and HILIC.
Monolithic Columns: Monoliths have shown promise but have been unable to displace the traditional microparticulate columns. The silica monoliths have been around for more than a decade and were based on the pivotal contributions of Tanaka and Nakanishi and coworkers (11). They were commercialized in the early 2000s by Merck KGaA, who licensed the patents from the Kyoto Institute of Technology in Kyoto, Japan. Tanaka and his research group have continued to study silica monoliths and have gained a better understanding of the physical and chemical properties. Because of the monoliths' small-sized skeletons and wide through-pores, much higher efficiency can be achieved than in the case of particle-packed columns at a similar pressure drop. Alternatively, first generation commercial silica monolithic columns have achieved efficiencies equivalent to a 4-µm silica column but with a pressure drop around 40–45% of the packed column run under the same linear velocity but the introduction of the second-generation monoliths may change their acceptance. Merck KgaA and its US affiliate EMD Millipore introduced the Chromolith HighResolution column at Pittcon 2012. The new columns display 50% higher efficiency than the first generation. Merck accomplished this by further optimization of the mesopore/macropore ratio of the silica monolith at the price of a slightly higher pressure than the first-generation monolith. However, since one of the attributes of the monoliths is their low-pressure drop, this increased pressure is not much of a disadvantage. Separations achieved with Chromalith HighResolution C18 endcapped rival those achieved with SPP columns using the same chromatographic conditions and column dimensions (12)albeit at a lower pressure drop. An improvement in tailing factors over the first-generation version, especially of basic compounds, also is a positive attribute of the second-generation monolith columns. The details of the second-generation silica monoliths are in the LCGC North America supplement (2).
Other advances in silica monolith columns include the introduction of capillary and microbore (2-mm i.d.) dimensions. One of the criticisms directed at monoliths was the high flow rates required to achieve separations comparable to packed-bed columns. The smaller internal diameter columns require lower flow rates and are more adaptable to mass spectrometry (MS). Still, the downside is that the longest length of commercial 4.6-mm i.d. silica monolith columns is 100 mm, but they can be connected in series to achieve higher plate numbers.
In the past, overall observations indicated that silica-based monoliths seem to work best for small molecules while the polymeric monoliths are best for macromolecules. More recently, reports have shown that polymeric monolithic materials can be synthesized that have better performance for small molecules (13). Some of the newer generation of high surface area "hyperlinked" polymeric monoliths should be more useful for small molecule separations. Using thermally initiated, free radical polymerization, monoliths also can be optimized for the separation of either small molecules or large biomolecules. The polymeric monolith phases have shown high mechanical robustness and low swelling in organic solvents because of their high level of crosslinking. Another approach to improving the performance of polymeric monoliths involves combining the technology of coating agglomerated latex charged ionexchange nanoparticles onto the surface of an oppositely charged monolith and using this material to separate small ions as well as large biomolecules. The thin layer of nanoparticles results in rapid mass transfer and therefore decent separation efficiency up to 50000 plates/m. Although few polymeric monoliths are currently on the market, the future could be bright if they are found to be reproducible, rugged and of high efficiency when manufactured on a commercial basis.
New Directions in Stationary-Phase Selectivity
Now that column efficiencies are approaching theoretical limits (reduced plate heights ~1.3–1.5), the future incremental gains in efficiency by manipulation of particle or monolith morphology may not provide dramatic improvements in chromatographic resolution. It has long been known that moderate improvements in selectivity can result in more gains in resolution (Figure 2). Thus, researchers are now investigating newer phases that might provide selectivity enhancements to tackle more difficult separations. There are four main areas where significant achievements in selectivity improvements have been made: HILIC, mixed-mode phases, supercritical fluid chromatography (SFC)–specific phases and specialty columns such as chiral columns.


Figure 2: Resolution and the effects of selectivity, efficiency and retention.
HILIC
In the last five years, a large number of HILIC phases have been introduced at Pittcon (12,14–17). HILIC is a separation technique for highly polar analytes that gets around some of the problems associated with reversedphase chromatography, such as low retention or phase collapse (dewetting). HILIC uses a polar stationary phase such as bare silica gel, a polar bonded phase (for example, diol), or certain mixed-mode or zwitterionic phases. Operating conditions usually require a high percentage of a nonpolar mobile phase, typically acetonitrile, similar to the requirements for normal-phase chromatography. However, unlike normal-phase LC, which uses nonpolar solvents like hexane and methylene chloride and tries to exclude water from the mobile phase, HILIC requires some water in the mobile phase to maintain a stagnant enriched water layer on the packing surface into which analytes may selectively partition. In addition, water-miscible organic solvents are used. With HILIC, polar analytes are well retained and are eluted in order of increasing hydrophilicity. HILIC is especially favoured by mass spectroscopists since ionization efficiency is often enhanced in organic solvents and the presence of low or no buffer salt compared to reversed-phase chromatography.
In our most recent HPLC column survey (18), HILIC usage doubled from the 2008 levels. Many users are discovering this useful mode to try out when reversed-phase HPLC doesn't quite do the job. Note that it is sometimes referred to as the aqueous normal phase mode, so don't let that confuse you.


Figure 3: LCâMSâMS separation of paroxetine and ranitidine in (a) reversed-phase and (b) HILIC mode (19). Reversed-phase conditions: Mobile-phase A: 8 mM ammonium formate in water; mobile-phase B: 8 mM ammonium formate in 95% acetonitrileâ5% water; flow rate: 0.3 mL/min; column temperature: 40 °C; sample volume: 5 µL. HILIC conditions: Same as for reversed-phase separation except column and gradient.
A simple illustration of how HILIC compares to reversed-phase chromatography can be seen in Figure 3. The analysis of drugs in biological fluids is an important area of focus in the pharmaceutical industry. In this example, we were looking to measure two drugs, paroxetine and ranitidine, spiked into serum (19). The sample preparation was simple. We used protein precipitation performed by the addition of acetonitrile followed by centrifugation. The supernatant liquid was directly injected into the LC–MS–MS system. Figure 3(a) shows the 10.5 min separation achieved by reversed-phase chromatography in which the more polar ranitidine elutes first with slight tailing on the C18 packing material. The paroxetine gave a fairly sharp peak. Gradient elution with an MS-compatible 8 mM ammonium formate buffer (A = weak solvent) and a 95:5 8 mM ammonium formate in acetonitrile–water (B = strong solvent) mixture was used to speed up the separation. Next, using a bare silica column of the same dimensions as the reversed-phase column and the same mobile phases, but with an opposite gradient (100–50% B in 10 min), we were able to separate the two compounds in just under 9 min, but the elution order was reversed [Figure 3(b)]. Both peaks displayed good peak shape. Using MS–MS detection, strong signals were observed even at the 0.5 ppb level (not shown) and the linear range was determined to be 0.5–100 ppb with r2 = 0.999.
There are several types of HILIC phases that are commercially available. The most popular are bare silica, RP-amide and zwitterionic phases (possessing two internal ionic functionalities in the bonded phase chain), but others with reversedphase and ion functionality, diol and multiple –OH groups, and different polar functional groups also have been used. Because an entire HILIC article is included in this supplement (6), I will not expand on the chemistries that are currently available.
Mixed-Mode Stationary Phases
Most separations are performed on straight phases where one main type of chromatographic interaction governs the separation. For example, when using reversedphase chromatography, organic compounds with an affinity for hydrophobic bonded phases will generally be retained by the order of increasing hydrophobicity. Other chromatographic interactions occurring at the same time will often confuse chromatographers and make separation behaviour different from one's expectations. On the other hand, it can be useful to provide multiple retention mechanisms if one has a particularly difficult separation where a single molecular interaction may not be enough to pull two similar compounds apart. To achieve a different selectivity, column researchers are now synthesizing phases that have two and even three different functionalities expressly for exploiting additional interactions and therefore increasing resolution by imparting different phase selectivity. In the last few years, mixed-mode phases have become popular and are helping to achieve separations that were more difficult only a few years ago.
For example, the simplest "mixedmode" phases may not really be another mode but different hydrophobic functionalities on the same packing. Phases such as phenyl-hexyl, C18PFP, C18-phenyl and even diphenyl have popped up and are being sold as alternatives to the popular C18. To illustrate something as simple as how a hydrophobic bonded group can affect a separation, consider the nine non-nutritive food additives separated isocratically in Figure 4 (20). Although C18 shows good retention for the compounds, lack of selectivity for the strongly retained additives precludes their separation. Merely replacing the C18 column with the phenyl-hexyl phase provides an excellent separation with a different elution order because of the differential attraction of the compounds to this phase.


Figure 4: Separation of non-nutritive food additives on two different reversed-phase columns (20): (a) Zorbax Eclipse Plus C18 and (b) Zorbax Eclipse Plus Phenyl-Hexyl. Column dimensions: 150 mm à 4.6 mm, 3.5 µm; mobile phase: 70:30 (v/v) 20 mM potassium phosphate (pH 2.5)âmethanol; flow rate: 1.5 mL/min; temperature: 25 °C; detection: UV absorbance at 220 nm. Peaks: 1 = ascorbic acid, 2 = saccharin, 3 = caffeine, 4 = p-hydroxybenzoic acid, 5 = aspartame, 6 = dehydroxyacetic acid, 7 = sorbic acid, 8 = benzoic acid and 9 = methylparaben.
More dramatic changes in functionality can lead to greater selectivity variations. When an ionic or ionizable functional group is added to a hydrophobic moiety, stronger interactions can occur. Popular pairings in this domain are C8-SCX (or WCX), C18-SCX (or WCX) and C18cationanion. Such groups will permit both hydrophobic and ionic interactions to occur simultaneously. For ionizable functional groups, pH can be used as a strong variable in optimizing separations. Quite a number of mixed-mode phases have been introduced in recent years (12,14–17).
Chiral Phases
Since coated polysaccharide patents have come to an end, a number of companies have entered the market with a variety of these popular phases (10–14). With the emergence of immobilized (bonded) polysaccharide chiral phases the use of a wider range of solvents and solvent combinations can be applied to cover the wide range of enantiomeric compounds. The Chiralpak IA and IB (Chiral Technologies) are immobilized versions of the Chiralpak AD and Chiracel OD. Chiralpak IC has a unique chiral selector, based on the 3,5-dichlorophenylcarbamate of cellulose, immobilized on 5-µm silica. Until now, these phases have not replaced the traditional coated polysaccharide phases for established applications. Apparently, the selectivities of the bonded chiral phases are different enough that they have not been a "drop-in" replacement for the coated polysaccharide phases. It is well known that a reduction of enantiomeric selectivity may occur when the chiral phases are fixed on the surface of a chromatographic packing. For a complete review of new chiral columns, refer to the article by Ward and Ward in this supplement (4).
Supercritical Fluid Chromatography (SFC)
Now in its third generation of instruments (21), SFC has been growing for the rapid separation of complex mixtures, especially for preparative work. Until now, SFC has been used mainly for the purification of chiral and other pharmaceuticals. The technique is very "green" because the main component of the mobile phase is carbon dioxide, which reverts to its gaseous state when the supercritical conditions are no longer present at the detector outlet. Supercritical carbon dioxide, which is relatively nonpolar, somewhat limits the types of compounds that can be successfully separated by SFC but with mobile phase additives, more polar analytes can be analysed. SFC users have been forced to use normal-phase HPLC columns such as bare silica gel, diol and cyano as stationary phases. Recently, companies have been developing stationary phases that are more suited to supercritical solvent mobile phases. Many of these phases are atypical and are relatively unknown to liquid chromatographers.
Only a few companies have delved into offering these unique SFC phases so far but as the market is growing for SFC, other companies will undoubtedly begin to offer suitable phases. Some popular phases that have been introduced and successfully used for SFC are 2- and 4-ethylpyridine, diethylamino, nitro- and dinitro-aromatic, aminophenyl and pentafluorophenyl. The websites of ES Industries, Princeton Chemicals, Phenomenex, Waters and a few other companies list many different commercially available phases with polar functionalities. The SFC columns have the same dimensions as HPLC columns so it is not a stretch for companies to enter this market provided they develop the bonding chemistries. For an update on columns for SFC, readers are directed to the recent article by Terry and Blair Berger (21).
Future Directions in HPLC Column Technology
Although silica gel and silica hybrids with chemically bonded phases still dominate the applications of HPLC, newer packing materials continue to be developed. So far, smaller particle sizes (less than 1.5 µm) of the conventional totally porous phases have not hit the market, although sub-2-µm superficially porous particles are available, but whether the current UHPLC instruments and columns can provide full performance is an open question. For those with relatively simple separations, today's high-throughput columns are quite adequate to provide rapid analyses of 1–2 min.
Whether the particle size reduction trend will continue is a matter of speculation. Smaller particles will require even higher pressure and place additional constraints on the packing materials, column hardware (especially frit design), instrument consumable parts like pistons, check valves and seals, and require increased attention to extracolumn effects and gradient delay volume. For those needing high resolution who want to use these smaller particles, the use of longer columns will place even more restrictions on the chromatography. It will be interesting to see if the superficially porous particle columns and the second generation of silica monoliths will slow down the "pressure race" because they offer great column efficiency at roughly half of the operating pressure.
Of course, another way to tackle the high-resolution problem is to employ multidimensional and comprehensive LC×LC techniques. Most workers in the field do not want to spend the time putting together an LC×LC system, but instruments to address this part of the market are becoming available. The question arises: How many people need to know and separate every compound in a complex mixture, perhaps consisting of hundreds or thousands of compounds? Often, only one or several sample components are of interest and a complex LC×LC system requiring multiple valves, tricky timing and hourslong separation times is overkill. But biochemists looking for one or two biomarkers in a complex bioassay may still need this sort of resolution power.
With plenty of plates to use for highefficiency requirements, users are beginning to turn their attention to using selectivity to bring about improved resolution of difficult compounds. It can be safely stated that reversedphase chromatography will continue to dominate applications and column introductions at Pittcon (12,14–17) as it has for the last three decades. With more than 1500 reversed-phase columns available in the marketplace (8), chromatographers have a large selection to deal with and the decision of which one to use is a matter of availability and past experience.
With the much wider availability of HILIC or aqueous normal phase columns, this technique should receive more attention from users who have faced the analysis of polar analytes unretained or poorly retained by regular reversed phase columns and don't want to turn to either ion-pair chromatography or normal-phase chromatography, with all of their gradient equilibration problems. Wider-pore HILIC columns are becoming available and may be useful in separating hydrophilic highermolecular-weight biomolecules or surfactants.
Sub-2-µm SEC columns are now available, but users are cautioned not to expect much smaller particle sizes of wide-pore packings where particle stability comes into play. More inert, biocompatible SEC columns would be appreciated for hydrophobic polypeptides. Separations of two classes of biomolecules, oligonucleotides and monoclonal antibodies, seem to have generated a lot of interest as bio-based pharmaceuticals move through the drug-development process. Certainly, columns for such separations have been quickly coming to market. Ionexchange or ion chromatography columns are almost 100% polymeric, which seems to hold up to the rigorous mobile phase and temperature conditions used for separations in this mode. More mixed phases will continue to be available for tough reversed-phase and HILIC separations. It will be interesting to see if the immobilized chiral chromatographic phases will take over new method development where both reversed-phase and normal-phase separations can now be performed with the same stationary phase.
References
(1) K. Wyndham, T. Walter, P. Iraneta, B. Alden, E. Bouvier, C. Hudalla, N, Lawrence and D. Walsh, LCGC Europe 25(10), 15–20 (2012).
(2) K. Cabrera, LCGC No. America 30(S4), 30–35 (2012).
(3) J.O. Omamogho, E. Nesterenko, D. Connolly and J.D. Glennon, LCGC Europe 25(10), 31–34 (2012).
(4) T.J. Ward and K.D. Ward, LCGC Europe 25(10), 28–30 (2012).
(5) H.G. Barth and G.D. Saunders, LCGC Europe 25(10), 21–27 (2012).
(6) E. Pontén, LCGC No. America 30(S4), 38–42 (2012).
(7) R.E. Majors, LCGC No. America 26(S4), 10–17 (2008).
(8) R.E. Majors, LCGC No. America 28(S4), 7–17 (2010).
(9) J.J.Kirkland, Anal. Chem. 64, 1239–1245 (1972).
(10) F.Gritti and G. Guiochon, J. Chromatogr. A 1221, 2–40 (2012).
(11) N. Tanaka, H. Kobayashi, N. Ishizuka, H. Minakuchi, K. Nakanishi, K. Hosoya and T. Ikegami, J. Chromatogr. A 965(1–2), 35–49 (2002).
(12) R.E. Majors, LCGC No. America 30(4), 290–310 (2012).
(13) F. Svec, J. Chromatogr. 1217(6), 902–924 (2010).
(14) R.E. Majors, LCGC No. America 26(3), 238–253 (2008).
(15) R.E. Majors, LCGC No. America 27(3), 206–222 (2009).
(16) R.E. Majors, LCGC No. America 28(3), 192–210 (2010).
(17) R.E. Majors, LCGC No. America 29(3), 218–235 (2011).
(18) R.E. Majors, LCGC No. America 30(1), 20–34 (2012).
(19) T. Yoshida, K. Yamanaka, H. Kumagai and R.E. Majors, "Hydrophilic Interaction Chromatography (HILIC) Separation of Basic Drugs using MS/MS Detection", Agilent Technologies Application Note No. 5989-3761EN, September 8, 2005.
(20) A.E. Brooks and W.J. Long, "Selective Analysis of Non-Nutritive Food Additives Using Agilent ZORBAX Eclipse Plus C18, Eclipse Plus Phenyl Hexyl, Eclipse XDB-Phenyl, and StableBond SB-Phenyl Columns," Agilent Technologies Application Note No. 5989-9951EN, November 3, 2008.
(21) T. Berger and B. Berger, LCGC No. America 28(5), 344–357 (2010).
Ronald E. Majors is a senior scientist in the columns and supplies division at Agilent Technologies in Wilmington, Delaware, USA. He also is the "Sample Prep Perspectives" and "Column Watch" editor for LCGC Europe. Direct correspondence about this article should be addressed to LCGC Europe, 4A Bridgegate Pavilion, Chester Business Park, Wrexham Road, Chester, CH4 9QH, UK, or e-mail the editor, Alasdair Matheson, at amatheson@advanstar.com










Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.




New Study Investigates Optimizing Extra-Column Band Broadening in Micro-flow Capillary LC
March 12th 2025Shimadzu Corporation and Vrije Universiteit Brussel researchers recently investigated how extra-column band broadening (ECBB) can be optimized in micro-flow capillary liquid chromatography.

