Development and Validation of an HPTLC Method for Determination of Aflatoxin B1
LCGC North America
A high performance thin-layer chromatographic (HPTLC) method was developed for the determination of aflatoxin B1 in cereals.
A simple, sensitive, precise, rapid, high performance thin-layer chromatographic (HPTLC) method was developed for the determination of aflatoxin B1 in cereals. Validation was carried out in compliance with International Conference on Harmonization Guidelines. The samples were extracted first using 8:2 methanol–water and were purified further by solid-phase cleanup using a polymeric sorbent, not described previously. Optimum conditions for the extraction were investigated. Separation and quantification were achieved by HPTLC using a bifarious mobile phase of 8:92 (v/v) acetone–chloroform on precoated silica gel glass plates. Densitometric analysis of aflatoxin B1 was carried out in the fluorescence mode at 366 nm. The calibration curves were linear in the range of 0.8–4.8 ng.
Aflatoxins are a group of toxic secondary metabolites mainly produced by the fungi Aspergillus flavus, Aspergillus nomius, and Aspergillus parasiticus growing in a wide range of foods and feedstuffs. Aflatoxins B1, B2, G1, and G2 are the only ones that occur naturally and are significant contaminants of a wide variety of feeds (1). Aflatoxins, especially aflatoxin B1, are toxic and carcinogenic to many species of animals, including humans (2). Because the International Agency for Research on Cancer (IARC, Lyon, France) has classified aflatoxin B1 in Group 1 as a human carcinogen (3), many countries and regions have set the maximum tolerable limit of aflatoxin B1. In the European Union, aflatoxin B1 is regulated in grains with maximum residue levels (MRLs) of 2 ng/g (4). In China, the maximum tolerable limit of aflatoxin B1 is 4 ng/g.
A wide variety of analytical methods has been developed for aflatoxins since their discovery in the early 1960s. Many methods are available for the qualitative and quantitative analysis of the aflatoxins, such as thin-layer chromatography (TLC) (5,6), high performance liquid chromatography (HPLC) (7–10), and enzyme-linked immunosorbent assay (ELISA) (11–14). The serious disadvantage to TLC is the lack of quantitative accuracy because of the commonly used visual estimation technique. Although ELISA is a fast and sensitive method, false positive results may be obtained. Because of sequential injection of the samples and standards in HPLC, it is time-consuming and wastes a significant amount of solvent. Compared to the other methods, high performance thin-layer chromatography (HPTLC) offers many advantages:
- Several samples can be run simultaneously using a small quantity of mobile phase, thus reducing the time and cost per analysis.
- An HPTLC instrument is inexpensive and easy to control.
- The technique is extremely flexible (15–17).
The aim of this study was to develop an economic, accurate, simple, rapid HPTLC method for the analysis of aflatoxin B1 in cereals. The sample purification step was performed by solid-phase extraction (SPE). The proposed method was validated with International Conference on Harmonization (ICH) guidelines (18,19).
Experimental
Reagents and Chemicals: Aflatoxin B1 was kindly supplied by Sigma Aldrich Chemie (Steinheim, Germany). All of the solvents were of HPLC grade and were obtained from Tedia Co. (Fairfield, Ohio). SPE cartridges were purchased from Dikma Technologies (Beijing, China).
Preparation of Standard Solutions: Aflatoxin B1 stock solution (0.5 mg/L) was prepared in 98:2 (v/v) benzene–acetonitrile. The standard solutions (40, 80, 120, 160, 200, 240 µg/L) were prepared by transferring 4–24 mL aliquots of stock solution to 50-mL volumetric flasks, and the flasks were filled to the mark with 98:2 (v/v) benzene–acetonitrile.
Preparation of Sample Solution: Cereal powder (20 g) was macerated with 100 mL of 80% methanol aqueous solution, and then underwent ultrasonic extraction for 10 min. Extracts were filtered immediately after extraction through folded filter paper; a portion (50 mL) was evaporated to dryness and the residue was dissolved in 10 mL of 10% methanol aqueous solution. The solution was passed through the ProElut PLS cartridge (Dikma, Beijing, China), and the flow was kept at 4 mL/min. The cartridge was allowed to dry for several minutes using vacuum to remove excess solution. The analytes retained were eluted with two 3-mL volumes of methanol. The effluents were combined and dried under nitrogen gas steam at 40 °C, and the residue was redissolved with 1 mL of 98:2 (v/v) benzene–acetonitrile.
Instrumentation and Chromatographic Conditions: Chromatography was performed on 20 cm × 10 cm glass plates precoated with 200-µm layers of silica gel 60 F254 (Merck, Darmstadt, Germany). Before analysis, the plates were cleaned by predevelopment with methanol and activated at 110 °C for 30 min. Samples were applied to the glass plates as bands by means of a Linomat 5 sample applicator (Camag, Muttenz, Switzerland) with a 20-µL volume, 2-mm bandwidth, 15-mm distance between the middle of the bands, and 1 µL/10 s delivery rate.
A Camag twin-trough chamber was saturated for 30 min at room temperature (25 ± 2 °C) with the mobile phase containing 8:92 (v/v) acetone–chloroform. After chamber saturation, the plates were developed to a distance of 70 mm. Subsequent to development, the plate was dried using a hair drier.
Densitometric scanning was performed using a Camag TLC Scanner III in reflectance fluorescence mode at 366 nm for all measurements and operated by the CATS software, version 1.3.4. The slit dimensions were 2 mm × 1.2 mm, and scanning speed was 20 mm/s. Spectral scanning was done over the range of 200–400 nm at a rate of 20 nm/s and data resolution of 100 µm/step.
Results and Discussion
Because aflatoxin B1 is soluble in moderately polar solvents, it is normally extracted using a mixture of organic solvents such as acetonitrile, chloroform, and methanol. It was reported that a mixture of methanol–water and acetonitrile–water provides good extraction efficiency (20–22).
Different extraction mixtures involving acetonitrile–water and methanol–water were examined for the extraction efficiency of cereal powder that had spiked with 10 ng/g aflatoxin B1 standards. Of the combinations investigated, 84:16 acetonitrile–water and 8:2 methanol–water are more suitable (Figure 1). Cereal powders were spiked with 5–15 ng/g aflatoxin B1 and extracted using 8:2 methanol–water and 86:14 acetonitrile–water. The results (Table I) show that 8:2 methanol–water provides better recovery.



Figure 1: Effect of extracting solvent mixture (A: methanolâwater and B: acetonitrileâwater) on the efficiency of extraction.
PLS (polymer sorbent) is an alternative sorbent that can be used for sample cleanup. Proult PLS contains a hydrophilic group and a hydrophobic group. Because it has better retention for polar and nonpolar compounds, it has a (HLB) hydrophile-lypophile balanced property. In this study, it exhibited very good retention capacity for aflatoxin B1 and removed fats, proteinaceous compounds, and other interfering compounds.


Table I: Recoveries (RSD [%]) of aflatoxin B1 spiked to soybean samples
The mobile phase composition was optimized by introducing small changes, and it was found that aflatoxin B1 was best resolved at Rf = 0.33 (Figure 2) in the solvent system of 8:92 (v/v)acetone–chloroform. The identity of the band of aflatoxin B1 in the sample extract was confirmed by overlaying its fluorescence spectrum with aflatoxin B1 standard using a TLC scanner III.


Figure 2: HPTLC chromatogram of aflatoxin B1 in Chinese soybean matrix (upper) and common nandina matrix (lower).
Validation
Limit of Detection and Quantitation: The standard solutions were applied as the upper method, representing 0.8–2.4 ng aflatoxin B1. The calibration curve of peak area against concentration was plotted, the regression equation for aflatoxin B1 was Y = -302.8 + 10.8X, and the correlation coefficient (r) was 0.99915.
The limit of detection (LOD) and limit of quantification (LOQ) were calculated using LOD = 3.3 × N/B and LOQ = 10 × N/B, where N is the standard deviation of the peak area (N = 6) taken as a measure of the noise and B is the slope of the corresponding calibration curve. To estimate the LOD and LOQ, blank 98:2 (v/v) benzene–acetonitrile was spotted six times by the method described above. The LOD for aflatoxin B1 was 0.22 ng and the LOQ was 0.66 ng.
Precision: The variability of the method was studied by analyzing standard solution aflatoxin B1 on the same day and on different days (intra- and interday precision). The results are given in Table II, which shows the variation of the aflatoxin B1 at three different levels: of 0.8 ng/spot, 2.4 ng/spot, and 4 ng/spot. The repeatability of sample application is expressed in terms of RSD% with respect to peak area.

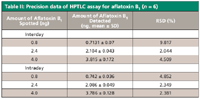
Table II: Precision data of HPTLC assay for aflatoxin B1 (n = 6)
Method application: In China, medicinal herbs have been used in the prevention, treatment, and cure of disorders and diseases since ancient times. However, aflatoxin contamination of medicinal herbs has attracted much attention (23–25). Following method optimization and validation, HPTLC was used to determine the contamination level of aflatoxin B1 in such Chinese medicinal herbs as magnolia vine fruit, indigowoad leaf, and common nandina. It also has good selectivity, high recovery and reproducibility (Figure 2). Figure 3 is a photograph of the developed plate.


Figure 3: Photograph of the developed plate. The first track (from left) is aflatoxin B1. The next tracks are aflatoxin B1 in different samples: sweet corn, peanut, Chinese soybean, fragrant rice, common nandina, magnolia vine fruit, and indigowoad leaf.
Conclusions
This article has described an HPTLC method developed for the identification and quantification of aflatoxin B1 in cereals. It can be used to quantify aflatoxin B1 in Chinese medicinal herbs by adjusting the proportion of the solvents in the mobile phase. The method is very useful for the rapid screening of different samples.
References
(1) B.S. Delmulle et al., J. Agric. Food Chem. 53, 3364–3368 (2005).
(2) M. Holcomb, C. Harold, and J. Thompson, J. Agric. Food Chem. 39, 137–140 (1991).
(3) J.W. Park et al., Food Addit. Contain. 19, 1073–1080 (2002).
(4) Commission Regulation (EC) NO.856/2005 of 6 June 2005 amending Regulation (EC) NO.466/2001 as Regards Fusarium Toxins (2005).
(5) M.W. Trucksess, W.C. Brumley, and S. Nesheim, J. Assoc. Off.Anal. Chem. 67, 973–975 (1984).
(6) J. Stroka, R.V. Otterdijk, and E. Anklam, J. Chromatogr. A. 904, 251–256 (2000).
(7) J. Blesa et al., Journal of Chromatography A. 1011, 49–54 (2003).
(8) W. Khayoon et al., Food Chemistry 118, 882–886 (2010).
(9) K. Reif and W. Metzger, Journal of Chromatography A. 692, 131–126 (1995).
(10) B. Giray et al., Food control 18, 23–29 (2007).
(11) S. Xiulan et al., International Journal of Food Microbiology 99, 185–194 (2005).
(12) E. Schneider, E. Usleber, and E. Märtlbauer, J. Agric. Food Chem. 43, 2548–2552 (1995).
(13) D. Saha et al., Anal. Chim. Acta. 584, 343–349 (2007).
(14) Barimehetoglu, O. Kuplulu, and T. Haluk Celik, Food Control 15, 45–49 (2004).
(15) A. Ghassempour et al., Chromatographia 64, 101–104 (2006).
(16) N. Singh et al., Chromatographia 63, 209–213 (2006).
(17) R. Dixit, C. Barhate, and M. Nagarsenker, Chromatographia 67, 101–107 (2008).
(18) ICH-Guidelines Q 2A, (CPMP/ICH/381/95) (1995).
(19) ICH-Guidelines Q 2B,Validation (CPMP/ICH/281/95) (1995).
(20) K.H. Otta, E. Papp and B. Bagocsi, Journal of Chromatography A. 882, 11–16 (2000).
(21) B. Huang et al., Analytica Chimica Acta 662, 62–68 (2010).
(22) C. Oliveira et al., Int. J. Mol. Sci. 10(1), 174–183 (2009).
(23) I. Rizzo et al., Mycotoxin Research 14, 46–53 (1998).
(24) I.P. Siu-Po and C.H.E. Chun-Tao, Journal of Chromatography A. 1135, 241–244 (2006).
(25) M. Ventura et al., Journal of Chromatography A. 1048, 25–29 (2004).
Hegang Gao, Li Chen, Guoshao Pan, and Chunyu Tu are with Shaoxing Center for Disease Prevention and Control, Zhejiang Province, China.















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).



