Determining Psychoactive Drugs in Blood Plasma and Serum Using Automated SPE–LC–MS/MS
A simple and rugged method to determine psychoactive drugs using automated SPE–LC–MS/MS is described.
The determination of psychoactive drugs is of great interest in the context of clinical and forensic toxicology. Easy and rugged determination of such compounds, down to the low ng/mL range from only 250 μL of blood plasma or serum, is described in this article. The comprehensively automated “multi method” is based on solid-phase extraction (SPE) coupled online to liquid chromatography with tandem mass spectrometry (LC–MS/MS). More than 60 compounds namely benzodiazepines, other sedatives, antidepressants, antipsychotics, methadone, and their relevant metabolites are included. The method has been fully validated for 52 compounds according to the guidelines of the German Society of Toxicological and Forensic Chemistry (GTFCh). Some less relevant drugs and metabolites are included for semi-quantitative or qualitative determination.
sychoactive drugs play a major role in many cases that are investigated in clinical or forensic toxicology laboratories. In addition to typical illicit drugs, such as, for example amphetamines, opioids, and cannabinoids, so-called new psychoactive substances (NPS) are often sold via the internet as legal highs, bath salts, or research chemicals. This trend has emerged in the last decade. Selling and purchasing these compounds is partly legal but legislative authorities have found ways of controlling these often dangerous substances by banning certain basic chemical structures.
In addition to illicit drugs, the abuse of prescription drugs dispensed for therapeutic purposes is an issue. Determining these compounds in blood plasma or serum and other biological matrices is the main topic of this article. Most prominent are the benzodiazepines, which are predominantly prescribed as sedatives, anxiolytics, and hypnotics and are the most widely used therapeutic drugs in psychiatry. Common for all benzodiazepines is the bicyclic structure consisting of a condensed diazepin- and benzene ring. Some well‑known active pharmaceutical ingredients are diazepam, lorazepam, and midazolam. Although benzodiazepines are less toxic than their predecessors, the barbiturates, intoxications can occur and can be fatal, especially in combination with other drugs such as alcohol and opioids. Long-term use can lead to physical addiction.
There are other sedatives that act in a similar way to benzodiazepines with a differing chemical structure referred to as non‑benzodiazepines or sometimes colloquially “z-drugs” since many of their names begin with a “z”. Like benzodiazepines they include nitrogen‑heterocyclic rings; typical representatives are zopiclone and zolpidem.
Beside tri- and tetracyclic antidepressants (TCA), the so-called selective serotonin noradrenalin reuptake inhibitors (SSNRI) are also prescribed as antidepressants. TCA are built of similar tri- or tetracyclic structures always containing a basic nitrogen atom, while SSNRI are a heterogeneous group of active pharmaceutical ingredients (API). Antipsychotics are drugs used for therapy of psychosis-like delusions, hallucinations, or paranoia. These are also a rather heterogeneous group of compounds.
In the context of prescription drugs methadone is also of interest. It is given in opioid replacement therapies under medical supervision. Co-consumption of sedatives and other prescription drugs is observed under replacement therapy in patients, to further dampen potential withdrawal symptoms. Co-consumption may need to be monitored to assess therapeutic success, which is one possible application of the analysis method described in this article.
Cases of driving under the influence of drugs (DUID) are another major area for which psychoactive compounds should be determined in blood. Driver impairment can be ascertained and causes of accidents can be elucidated. Moreover intoxications in clinical or forensic context can be discovered. When these substances are used in drug‑facilitated crimes and, for example, given as “knockout drops” the victims’ blood must be examined.
A look into the scientific literature (1–7) reveals that benzodiazepines and other prescription drugs are normally extracted by liquid–liquid extraction (LLE) or solid‑phase extraction (SPE) from serum or plasma, oftentimes preceded by a protein precipitation step. Sometimes solid‑supported LLE or solid-phase microextraction (SPME) are employed. Resulting extracts are routinely analyzed by liquid chromatography tandem mass spectrometry (LC–MS/MS) or, after a derivatization step, by gas chromatography (GC)–MS/MS. With the availability of more sensitive mass spectrometers, protein precipitation without further cleanup or enrichment has been used for LC–MS/MS determination of the aforementioned compounds.
The comprehensively automated SPE–LC–MS/MS method described in this article was developed at the Institute of Legal Medicine in Cologne, Germany, based on a manual SPE workflow from the Institute of Legal Medicine in Muenster, Germany.
Experimental
For automation of the sample preparation steps and injection of the final extract into the LC–MS/MS system a MultiPurpose Sampler (MPS, Gerstel) was employed. The automated system consisted of a Dual Head MPS (Gerstel) with a 2.5-mL syringe on the left head for all sample preparation steps and a 100-μL syringe for injection of the prepared sample extract into the
LC–MS/MS (1200 SL LC and 6460 MS, Agilent Technologies). Modules for SPE, extract evaporation, and solvent reservoirs were adapted to the autosampler.
Initially a 250 μL serum or plasma sample was added and mixed with 25 μL of an internal standard (ISTD) solution in a 2-mL vial. In the case of a hemolytic or post‑mortem specimen a protein precipitation step with acetonitrile was performed before the SPE. Sample vials were then placed in the autosampler and all further preparation steps, including injection to the analysis system, were executed automatically. Since all the included compounds can be protonated at acidic pH values, a cation exchange sorbent was chosen for analyte enrichment and cleanup (Chromabond HR-XC, 60 mg, 45 μm, 3 mL format, REF 730956P45MPS, Macherey-Nagel). The SPE automation relies on standard SPE cartridges that are cut and equipped with a transport adapter at the top and a disposable canula on the Luer port at the bottom. The cartridge was conditioned by injecting each 1 mL of methanol, water, and phosphate buffer pH 6. The sample was diluted with 1 mL of phosphate buffer pH 6, loaded onto the SPE cartridge, and analytes were bound by the nonpolar backbone of the sorbent material followed by washing steps with 1 mL phosphate buffer and 70:30 (v/v) water–methanol. During these steps, medium‑polar and polar interferences were washed away. In the next step the cartridge was washed with 0.1 M HCl protonating the analytes and binding them to the cation exchange groups of the sorbent. A sequence of washing steps, including methanol, followed in order to further remove interfering compounds bound to the nonpolar backbone of the sorbent while the analytes were bound to the cation exchange groups. In between these steps and directly before analyte elution, the cartridge was dried in a stream of nitrogen delivered through the 2.5-mL syringe, which was connected to a gas supply. Compounds of interest were eluted two times with 500 μL of a mixture of ethyl acetate and ammonia. The eluates were collected in vials fitted with septum caps and were subsequently evaporated to dryness in the evaporation module for 10.5 min at 50 °C under shaking and reduced pressure comparable to a rotary evaporator. The extracts were reconstituted in 100 μL of a 85:15 (v/v) water–acetonitrile mixture containing 0.1% formic acid. A 30 μL aliquot of this solution was injected into a 5 μL sample loop for transfer onto the LC column, a 150 × 2 mm, 2.7 μm Nucleoshell RP 18 (Macherey‑Nagel). Analytes were separated during a 15-min gradient run with a flow of 0.3 mL/min employing water and acetonitrile, both including 0.1% formic acid, and detected in dynamic multiple reaction monitoring mode (MRM). For each target compound two MRM transitions were chosen, one quantifier and one qualifier, in addition to one MRM transition for deuterated internal standards (ISTD) used for quantification.
Results and Discussion
The analysis method was completely validated according to guidelines of the GTFCh (8) for 52 of the 65 target analytes (Table 1). Seven further compounds were considered as “semi‑quantitative” because the complete validation data had not yet been collected. For an additional six compounds a “qualitative” detection can be performed by having established a cut‑off value, which includes evaluation of extraction recovery and matrix effects. With regard to “semi‑quantitative” and “qualitative” compounds, quantitation is not required on a regular basis at the laboratory in Cologne.


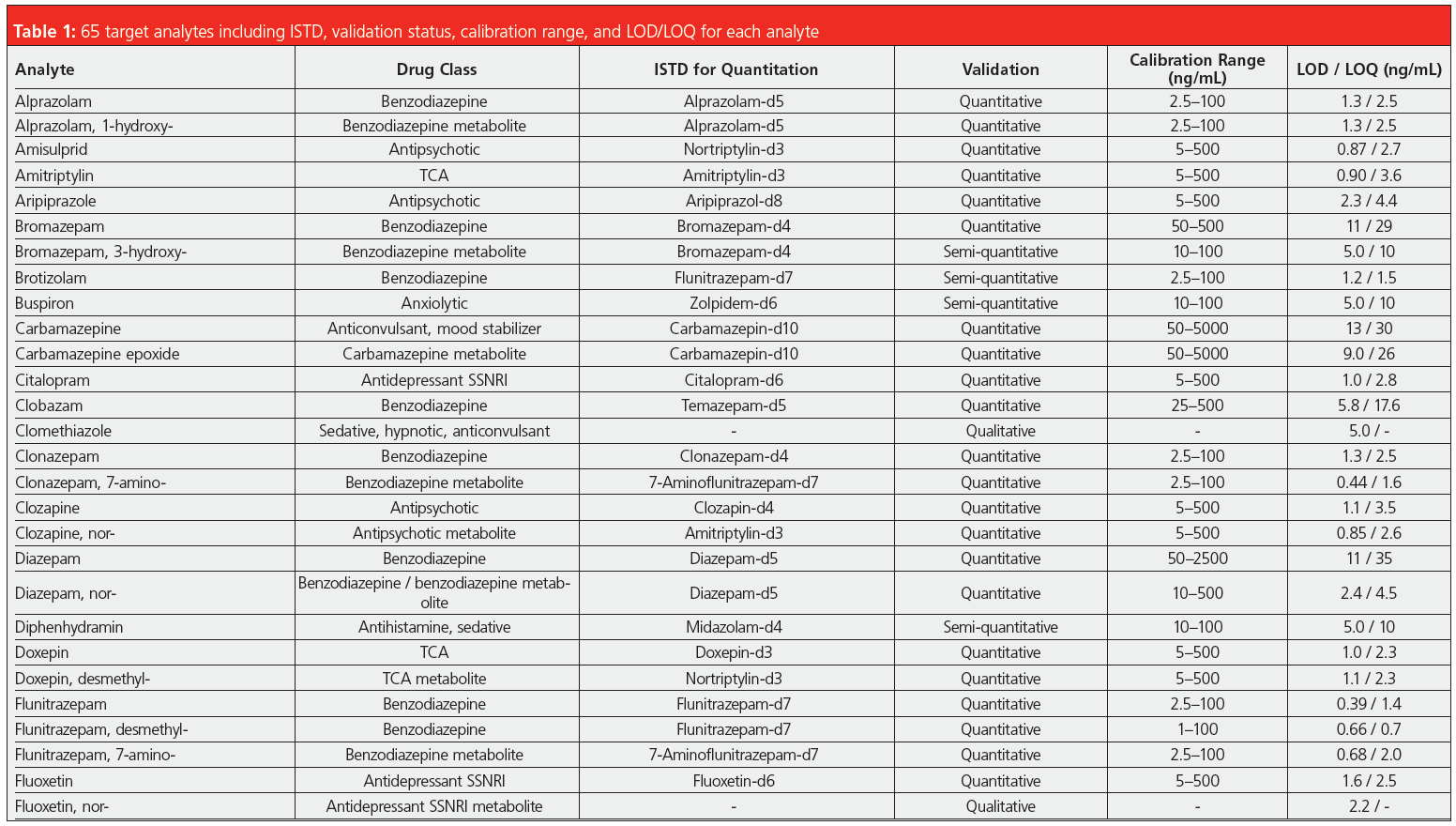

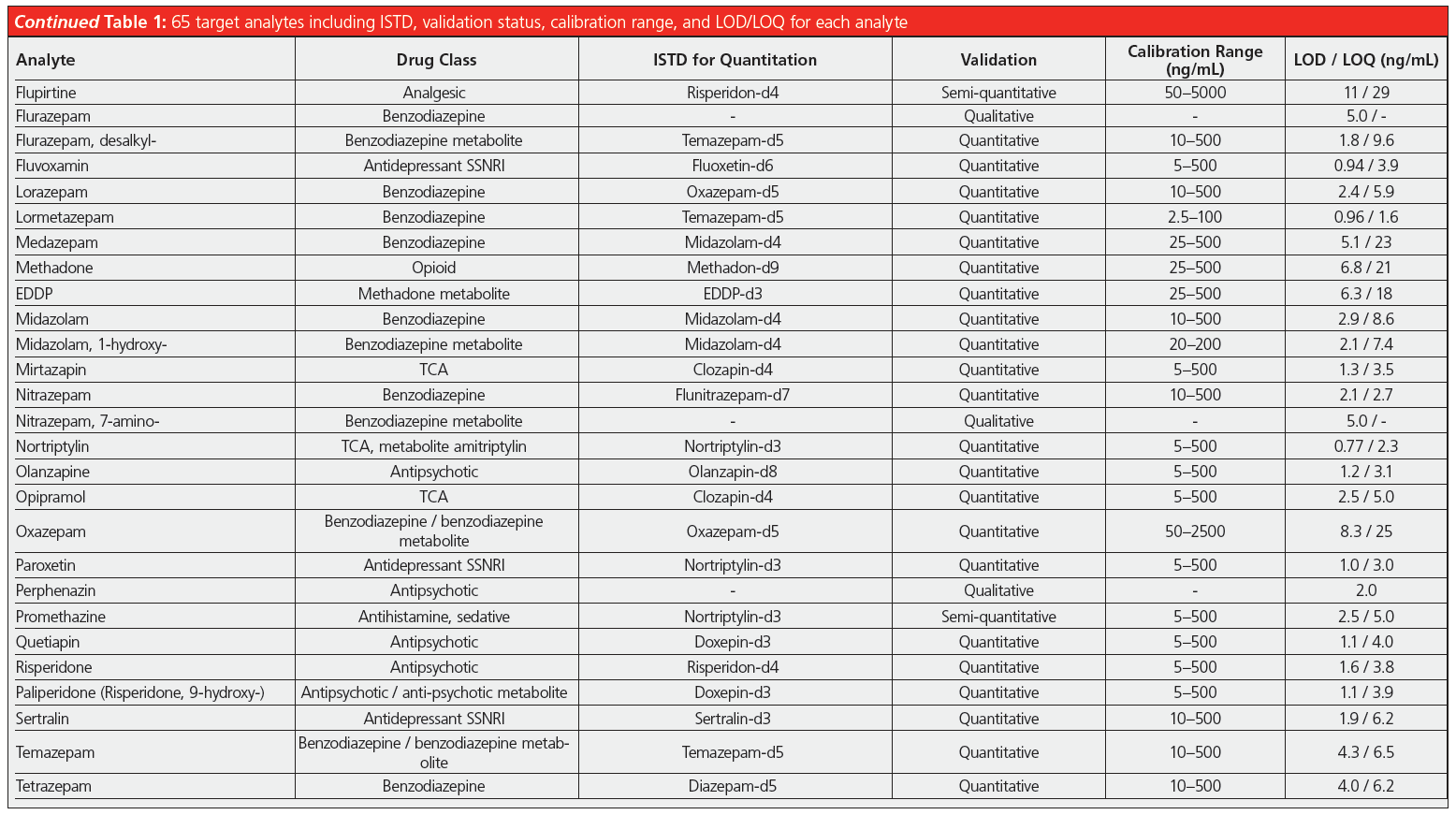

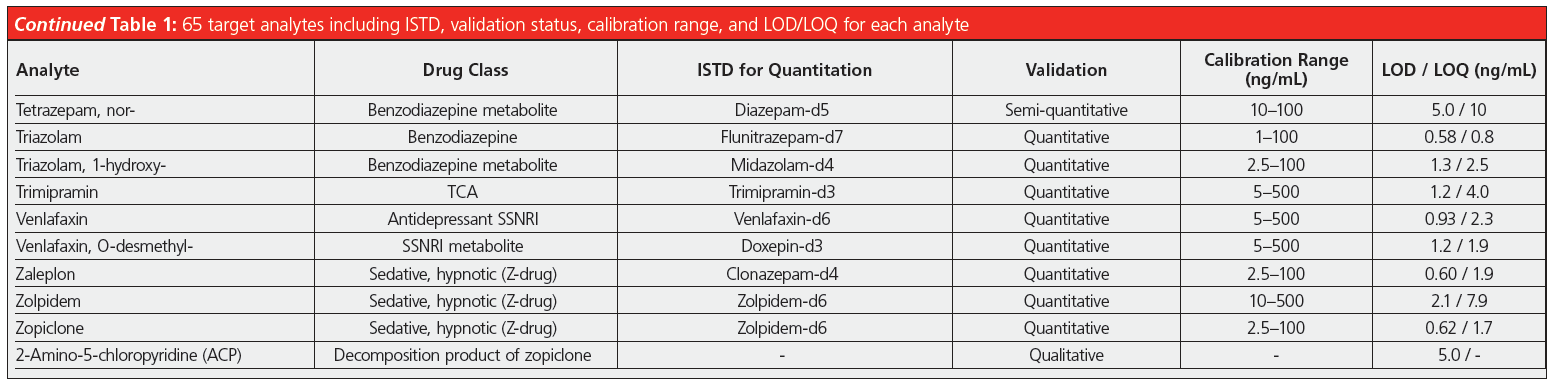
Validation comprised establishing limits of quantification/detection (LOQ/LOD) and the linear range. Furthermore, precision (repeatability and time‑different intermediate precision) and accuracy were tested at low and high concentrations inside the linear range as well as the extraction recovery and matrix effects. Limits of quantification were between 1–35 ng/mL, and were below 10 ng/mL for the vast majority of compounds. The linear ranges reached upper values between 100–2500 ng/mL. For practical reasons, in many cases the lowest calibration point was chosen slightly above the LOQ to facilitate the preparation of calibration solutions. Precision data expressed as relative standard deviation were mainly below 8% and often even below 5%, proving the excellent repeatability of such an automated analysis method. Spiked blank matrix was used for calibration, blank matrix spiked with internal standard only as matrix blank sample, and external quality control samples were analyzed in every sequence.
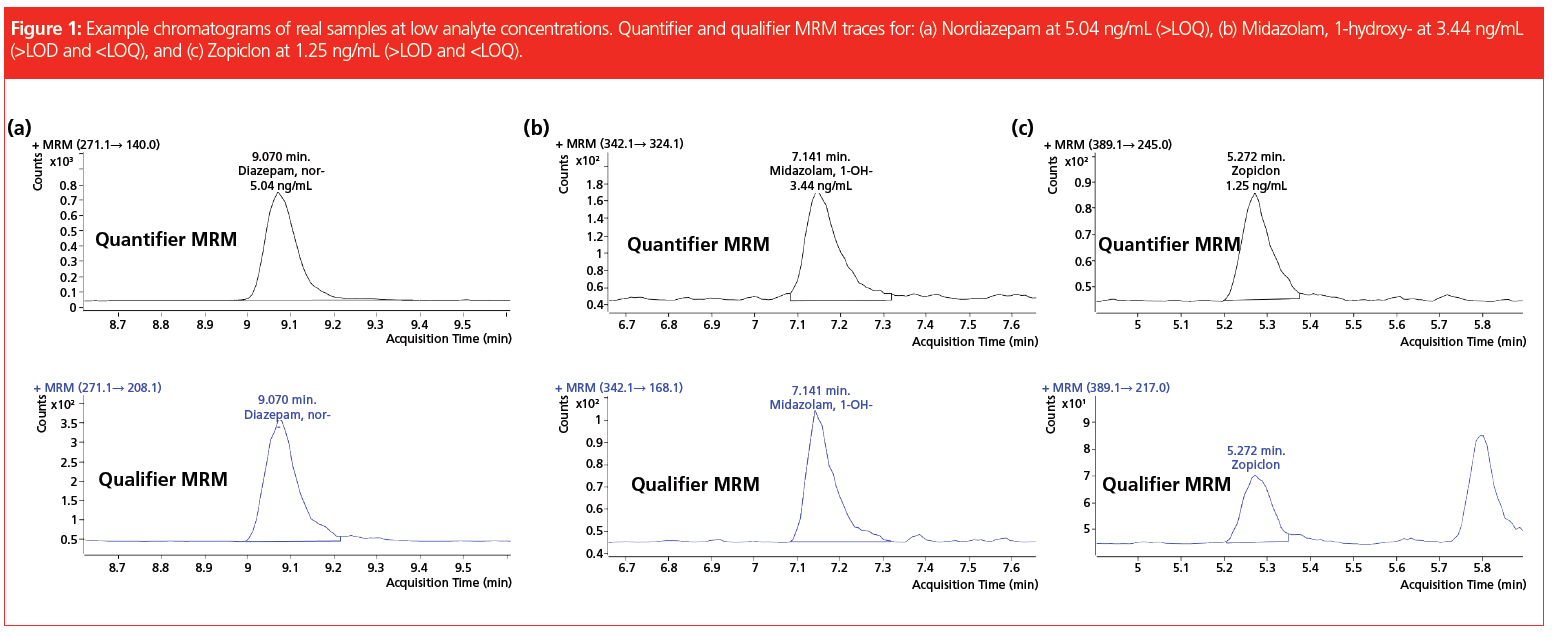
In addition, the laboratory successfully took part in available “round robin” tests for the following compound classes: benzodiazepines, opioids, z-drugs, tricyclic antidepressants, and selective serotonin noradrenalin reuptake inhibitors. The developed method is included in the DIN EN ISO/IEC 17025:2018 accreditation of the forensic toxicology laboratory at the Institute of Legal Medicine in Cologne. It is employed routinely for serum, plasma, whole blood, and post‑mortem specimens, adding up to roughly 1500 samples analyzed so far. Since a couple of compounds cannot be detected by immunochemical pre‑testing, the described SPE–LC–MS/MS method is used as a multi-targeted screening that includes confirmation analysis and quantitation, if necessary. Before implementation of this method the parameters were contained in two separate workflows running on a standalone SPE extractor. Each of them needed 0.5 mL of sample resulting in a total required volume of 1 mL, while today only 0.25 mL is needed for the complete set of analytes. The former methods demanded supervision and several manual interventions, such as evaporation of the extracts and reconstitution, which required the undivided attention of laboratory personnel, making it difficult to perform other tasks. Comprehensive automation of the workflow, including LC injection, reduces the workload and gives laboratory personnel time for other important tasks, such as data review or developing new analysis methods. Due to the extensive cleanup and enrichment procedure, good quality chromatograms with very low levels of interfering compounds are achieved (Figure 1) and the maintenance effort for the LC–MS/MS instrument is reduced. Analyte peaks can be identified unambiguously and automated integration results in accurate quantification without the need for reintegration for the vast majority of peaks.

Conclusions
This comprehensive automation of complex sample preparation workflows, including the injection into an analysis system, greatly reduces the manual workload. Data quality is high without depending on manual intervention and reintegration. In addition to the application described in this article, the laboratory in Cologne also employs a similar setup coupled to a GC–MS/MS system for the automated determination of cannabinoids and metabolites in serum and hair. These examples illustrate the benefits of implementing automated sample preparation methods into laboratory workflows.
Acknowledgements
The author would like to thank Clara Reinartz, Tobias Kieliba, Maren Fussberger, and Hilke Andresen-Streichert Ph.D. from the Institute of Legal Medicine in Cologne, Germany, for carrying out the major part of method development, the validation, and for sharing all relevant information.
Jennifer Schuerenkamp Ph.D. from the Institute of Legal Medicine in Muenster, Germany, is acknowledged for the development of the initial manual analysis method.
References
- K. Persona, K. Madej, P. Knihnicki, and W. Piekoszewski, J. Pharm. Biomed. Anal. 113, 239–264 (2015).
- F.E. Dussy, C. Hamberg, and T.A. Briellmann, Int. J. Legal Med. 120(6), 323–330 (2006).
- I.I. Papoutsis, S.A. Athanaselis, P.D. Nikolaou, C.M. Pistos, C.A. Spiliopoulou, and C.P. Maravelias, J. Pharm. Biomed. Anal. 52(4), 609–614 (2010).
- F. Mußhoff and T. Daldrup, Int. J. Legal Med. 105, 105–109 (1992).
- D. Montenarh, M.P. Wernet, M. Hopf,
H.H. Maurer, P.H. Schmidt, and A.H. Ewald, Anal. Bioanal. Chem., 406, 5939–5953
(2014). - I.I. Miroshnichenko and N.V. Baymeeva, J. Chrom. Sci. 56(6), 510–517 (2018).
- E. Saar, J. Beyer, D. Gerostamoulos, and O.H. Drummer, Drug Test. Anal. 4(6), 376–394 (2012).
- Guidelines for quality control in forensic toxicological analyses, GTFCh 20090601-20180125. https://www.gtfch.org/cms/index.php/en/guidelines
Oliver Lerch works as Senior Application Scientist in the Analytical Services Department at Gerstel. After receiving his Ph.D. from the Ruhr University Bochum, Germany, in 2003, he worked at the Max-Planck‑Institute of Molecular Plant Physiology in Potsdam, Germany, before joining Gerstel in 2005 where he has been instrumental in the development of technologies and products. These include applied customer projects on automated GC–MS and LC–MS solutions.
E-mail: Oliver_Lerch@Gerstel.de
Website: www.gerstel.com




















New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.


