A Decade of Quantitating Cyanide in Aqueous and Blood Matrices Using Automated Cryotrapping Isotopic Dilution Static Headspace GC-MS
Paul R. Loconto shares his insights into the principles and practice of implementing and improving upon the original method to quantitate cyanide at trace concentration levels in human blood matrices using automated cryogenic trapping isotope dilution static headspace gas chromatography-mass spectrometry (GC-MS).
Paul R. Loconto, Recently retired from the Michigan Department of Community Health, Bureau of Laboratories, Lansing, Michigan, USA.



The author shares his insights into the principles and practice of implementing and improving upon the original method to quantitate cyanide at trace concentration levels in human blood matrices using automated cryogenic trapping isotope dilution static headspace gas chromatography-mass spectrometry (GC-MS).

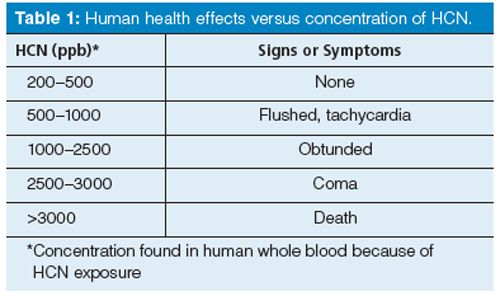
Often when one hears the word “cyanide”, concern and even fear arise. The pH of hazardous waste being stored must be monitored over time. If this waste becomes too acidic, toxic hydrogen cyanide (HCN) could be released into the atmosphere. Humans who are in close proximity to the stored hazardous chemical wastes might be exposed unknowingly. Table 1 correlates human blood concentration levels to potential health effects. Exposure to 150 ppm for 0.5-1 h may endanger life. Death may result from a few minutes of exposure to 300 ppm. The average fatal dose is 50-60 mg (1). Interest remains strong in the need to biomonitor and assess exposure to the toxic gas HCN from a public health perspective. Blood concentration levels in the general population are as high as 200 ppb, derived from dietary consumption of vegetables such as brussels sprouts (2). The cyanide ion (CN-) is the conjugate base of the weak acid HCN (pKa = 9.2). It is a moderate Brönsted-Lowry base. It is isoelectronic with carbon monoxide and nitrogen and thermodynamically very stable. CN- is found either coordinated to a transition metal ion or within an ionic lattice associated with an alkali or alkaline-earth metal ion. The alkali metals cyanide salts are considered the most highly toxic because of the ease with which HCN can evolve if these salts are acidified. The CN average bond enthalpy (ΔH°298 = 879 kJ/mol) in cyanide is relatively high because of the triple bond. Note that ΔH°298 = 293 kJ/mol for a C-N single bond (3). I have taken the opportunity, when equipped with a Fourier transform infrared (FT-IR) microscope, to place a small amount of KCN crystals on a slide and record the spectrum. A strong absorption band because of the triple bond at 2080 cm-1 is evident and diagnostic for the presence of cyanide in an inorganic compound (4). Quantitating CN- in drinking water samples is routine in environmental testing laboratories. I first observed the quantification of CN- in aqueous matrices using the chloramine-T/pyridine-barbituric acid colorimetric method. A review of most analytical approaches to detect CN- in aqueous and biological matrices cites recent studies using spectrophotometric, fluorometric, potentiometric, amperometric, and capillary electrophoretic, as well as gas chromatography-mass spectrometry (GC-MS) methods (5). The United States Environmental Protection Agency (US EPA) has recently promulgated a new method to quantitate CN- in drinking water matrices using the automated cryotrapping static headspace isotope dilution GC-MS technique (6). Recent interest in using various cobalt complexes related to vitamin B12 to quantitate CN- spectrophotometrically is growing and follows the initial work of Männel-Croisé and colleagues (7). A specific complex of vitamin B12, a hydroxoaquocobinamide, was used to develop a method to quantitate CN- spectrophotometrically in blood matrices (8). Quantitating CN- in blood matrices using automated cryotrapping isotopic dilution static headspace GC-MS occupied significant amounts of my time over the past decade. With strong support from the United States Centers for Disease Control and Prevention (CDC) and starting with a few laboratories over a decade ago, there are now several dozen state and municipal public health laboratories (SPHLs) in the United States that have a surge capacity capability to provide trace level environmental health monitoring to quantitate CN- in whole blood using this GC-MS approach. This important and significant investment in analytical chemistry capacity is warranted since sodium and potassium salts containing cyanide are easily obtained and abused, thus posing a public health threat. In this article, I introduce important theoretical underpinnings to help readers better understand how automated cryotrapping static headspace isotope dilution GC-MS is used to find the concentration of CN- at trace levels in whole blood matrices. I discuss the unique acid-base chemistry of the cyanide ion initially dissolved in an aqueous matrix. I discuss the significance of a recent paper that establishes the effect of temperature on the Henry’s law constant for partitioning dissolved HCN between sample and headspace. Control of temperature is of utmost importance to headspace sampling using the automated cryotrapping static headspace isotope dilution GC-MS technique to quantitate CN- in whole blood. I review a more fundamental approach to isotope dilution and show how calibration could be eliminated in quantifying CN-. Finally, I discuss practical aspects of a decade-long approach to improving reagent additions to the blood sample, sample sequence programming, HCN peak shape via changes in GC inlet parameters, and porous layer open tubular (PLOT) column performance. I also discuss the influence of isobaric interference when using an isotopically labelled internal standard with chemically deteriorated blood samples.
Experimental
Instrumentation: A model 6890/5973N GC-MSD system (Agilent Technologies) that incorporates a split-splitless inlet with electronic pressure control and a mass-selective detector was used to generate the data. The GC system is interfaced to a workstation that uses ChemStation (Agilent) software for chromatographic control, data acquisition, and storage. An MPS2 autosampler Gerstel GmbH & Co KG) was also used that consisted of a dualâsyringe delivery system, located on top of the GC-MS system. Additional accessories facilitate all other aspects of reagent delivery to the sample including sample agitation and incubation at a fixed temperature, headspace sampling, and injection into a split-splitless inlet. The temperature of the headspace syringe can be set and the needle flushed with inert nitrogen gas after injection. Software designed to operate the autosampler, Maestro (Gerstel), was integrated within the ChemStation software. A CIS4 inlet (Gerstel) located in the rear and a CTS2 cryotrap (Gerstel) are factory installed on newer instruments. The instrument must be equipped with a means to cryotrap the analyte downstream from the column inlet. A liquefied nitrogen Dewar flask that includes an insulated gas line provides cryogenic cooling to the cryotrap at -30 °C. The headspace syringe waits for a ready signal from the cryotrap controller before it will collect the sample and inject. A 1.0-mL aliquot of headspace was sampled and injected into the splitless inlet. The PLOT column temperature was programmed as follows: 100 °C to 120 °C at 4 °C/min then heat rapidly to 200 °C. Helium was used as the carrier gas at a flow rate of 1.0 mL/min. The chemical composition of the headspace consists of acidic moist air that contains a trace of HCN. The ready signal will not appear until the cryotrap is to within 1 °C of the cryotrapping setpoint after equilibration has been attained.
Automated Reagent Addition and Sample Introduction:
The operational aspects of the method have evolved over the past decade. The method uses the autosampler’s upper tier rail to move a liquid handling syringe head mount and the lower tier rail to move a headspace syringe head mount. The automation steps for the current method are as follows: 1. A precise 100-µL aliquot of the internal standard reagent (K13C15N in 0.1 N sodium hydroxide at partsâper-million levels) is added from a 10-mL reservoir to the first headspace vial in the sample sequence; 2. A precise 250-µL aliquot of mixed acids reagent (50 mg ascorbic acid/mL in dilute phosphoric acid (between 5-50% phosphoric acid diluted from concentrate) is added from a 10-mL reservoir to the first headspace vial in the sample sequence; 3. Steps 1 and 2 will be repeated for each and every headspace vial in the sequence with syringe rinsing after the last vial; 4. The first headspace vial will be transported to the heated agitator and pulse agitated for 4 min with the headspace syringe at 65 °C and the agitator at 60 °C; 5. The first vial is headspace sampled at 65 °C and 1.0 mL of headspace is injected into the GC system; 6. Steps 4 and 5 will be repeated for each and every headspace vial in the sample sequence.
Blood Specimen Storage and Trace Quantitative Analysis:
If the samples arrive in the laboratory in vacuum blood collection tubes (Vacutainer, Becton Dickinson), 0.5 mL of the blood specimen is transferred to a 10-mL headspace vial that is then capped and stored at 4 °C for subsequent analysis. If the frozen sample arrives to the laboratory in 10-mL headspace vials, the analyst should place the samples in a freezer at -20 °C until the analysis is to occur. Chain of custody protocols are followed. The sample is then removed from the freezer and allowed to thaw. The analyst needs only to replace the cap and septum with one more suitable to puncture by a headspace syringe and store at 4 °C for subsequent analysis. Automated addition of a strong acid such as H3PO4 to blood samples that have been previously enriched with isotopic CN- under alkaline pH conditions in a closed headspace vial completely converts all dissolved CN- to dissolved HCN in the closed headspace vial. Headspace sample vials await analysis at a chilled temperature via Peltier cooling at 5 °C so as to maintain sample integrity. One autosampler sample tray can accommodate up to 32 10-mL headspace vials. The analytes from the blood specimen are quantitated: H12C14N, m/z 27 (quantitative ion); 12C14N, m/z 26 (qualifier ion); and its isotopic analog H13C15N, m/z 29 (internal standard); and are cryotrapped at a location on the PLOT column nearer to the inlet. The cryotrap is rapidly heated to thermally desorb the analytes. Analytes of interest are chromatographically separated from interferences. H12C14N and H13C15N are quantitated using GC-electron ionization (EI)-MS with selected ion monitoring (SIM) under programmed temperature conditions. The retention times for H12C14N and for H13C15N are, of course, identical. Calibration and quantification is based on isotope dilution. This method was pioneered at the CDC (9), then transferred to the SPHLs. This method was subjected to further study at the National Institute of Standards and Technology (NIST) (10). The method has served the needs of the Laboratory Response Network - Chemical (LRN-C) over the past decade.
Results and Discussion
CN- as a Moderately Strong Base in Water-HCN Partitioning into Headspace: The moderately strong Brönsted-Lowry base, CN-, with a Kb = 1.6 × 10-5 (11) reacts with water and equilibrates in aqueous solution according to equation 1:

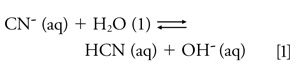

Figure 1: Fraction of cyanide as either HCN or CN- as a function of solution pH.
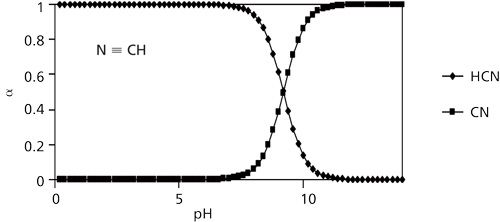
The pH dependence on the relative distribution of HCN and CN- is critical to successfully quantitating HCN in aqueous or blood samples. To store aqueous solutions that maintain a CN- concentration that is unchanged over time, the pH should be quite alkaline. Using equations 2 and 3 (below) I show that at a pH of 11.4, more than 99% of all cyanide is in the form as CN-. Adding OH- drives the above equilibrium to the left, suppressing the hydrolysis of CN-. This eliminates any possible analyst exposure to HCN. Equations 2 and 3 show how the fractions of HCN and CN- depend on Ka and [H+]. αHCN and αCN- are plotted as a function of aqueous solution pH in Figure 1.

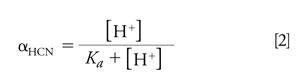

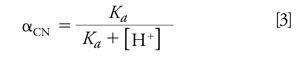
CN- exists as either free when dissolved in water or chemically bound to inorganic and organic metal complexes. Upon acidifying, OH- is removed as it reacts with excess added H+ to form H2O. This shifts the equilibrium to the right in favour of forming HCN. Applying equation 2 shows that, at a pH of 6.2, 99.99% of all cyanide is HCN. If the matrix pH is acidified using strong acids such as phosphoric acid, a stoichiometric amount of HCN is formed from cyanide. HCN (aq.), once formed in an acidic solution in a closed headspace vial, partitions into the headspace in accordance with Henry’s law for dissolved gases in water. A linear relationship exists between HCN’s vapour pressure and its aqueous concentration (applicable only to dilute solutions) at a fixed temperature and this relationship between a solute’s vapour pressure and its concentration was first observed and reported in 1803 by William Henry (12). The proportionality constant depends on temperature and is known as the Henry’s law constant. The application of the Henry’s law constant is frequently used in environmental chemistry problem solving (13). A recent contribution to the literature provides a review of earlier experiments to find the Henry’s law constant for HCN and reports new experimental values (14). Effluent gaseous HCN in equilibrium with constant temperature aqueous CN- was collected in dilute sodium hydroxide and determined spectrophotometrically using the hydroxoaquocobinamide complex discussed earlier. These temperatures cover the physiologically important temperature range of 18 °C to 38 °C while generating trace levels (parts per billion to parts per million) of HCN. The vapour-solution equilibrium of HCN was determined in 0.1 M sodium phosphate buffer adjusted to pH 9.00. Referring to Figure 1, at a pH of 9.0, αHCN equals 0.617 and αCN- equals 0.383. Hence ~62% of the available cyanide entered the gas phase in these experiments. The authors found that kH as a function of temperature for HCN may be expressed as

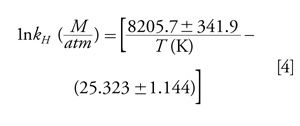

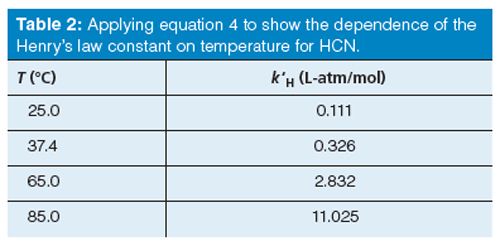
The inverse of kH (M/atm) is k′H (L-atm/mol). Table 2 lists values of k′H versus temperature using equation 4. The influence of temperature on k′H shows one and two order of magnitude increases in the partial pressure exerted by HCN in the headspace as the temperature increases from 25 °C to 85 °C. Recall that in our current method, the headspace syringe temperature is set at 65 °C and the incubation temperature within the agitator is set at 60 °C. As k′H increases with increasing temperature, increasing numbers of HCN molecules partition into the headspace. This leads to greater analyte sensitivity and, subsequently, lower method detection limits (MDLs). Lower MDLs for HCN in whole blood closely approximate the so-called baseline or background concentration level of HCN found in the blood of humans. The sample pH and temperature dependence of k′H are the two most significant factors that contribute to a highly sensitive analytical method. In contrast, temperature and pH play little to no role when optimizing static headspaceâGC parameters when volatile organic compounds are partitioned from aqueous matrices (15).

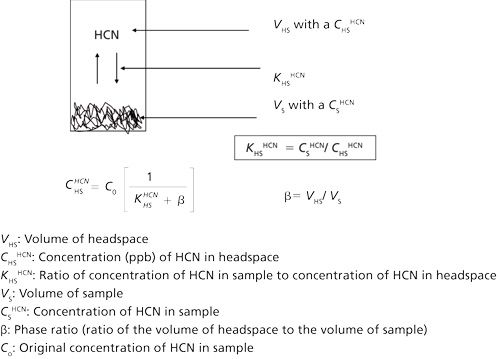
Figure 2 schematically shows the partitioning of HCN molecules into the headspace of a sealed vial while defining and depicting the important mathematical relationships based on the distribution coefficient KHSHCN between a condensed phase and the headspace at a fixed temperature. Analytes that more effectively partition into the headspace from the condense phase have lower KHS values than analytes that do not. For example, at 50 °C, KHSethanol is 511 while KHS n-hexane is 0.043 (15). The integrated area under a chromatographically resolved peak for HCN is directly proportional to its concentration in the headspace: AHCN ∞ CHSHCN. The concentration of HCN in the headspace in turn can be related to the original concentration of HCN in the blood sample according to the following relationship: CHSHCN = Co[1/(KHSHCN + β)] [5] Equation 5 forms the basis of quantification and shows why the distribution coefficient and the phase ratio must be kept constant in all calibration standards, quality control (QC) standards, and unknown samples. I have found that a precise and accurate sample volume must be taken (0.5 mL of aqueous solution or 0.5 mL of whole blood in this method). Placing exactly 0.5 mL of sample into the 10-mL headspace vial avoids any systematic error.
Calibration for HCN Could Be Eliminated (Isotope Dilution in Its Purest Form):
A fixed amount of isotopically enriched cyanide (K13C15N, internal standard) is added to both calibration standards and samples before headspace sampling. The ratio of the MS abundance of 1H12C14N (native) to the MS abundance of 1H13C15N (labelled) versus concentration of HCN (native) in parts per billion establishes the calibration while interpolation of this ratio for a sample results in HCN quantification. Calibration is conducted to compensate for small impurities in the analyte and internal standard (16). Quantitative analysis of blood specimens due to human exposure to HCN can be estimated without the need for calibration. Fassett (17,18) has provided the mathematical relationships to do this:


Equation 6 assigns a 1 to the isotope of HCN with the lower mass number and assigns a 2 to the isotope of HCN with the higher mass number. The terms in equation 6 are defined as Rm is the experimentally measured abundance ratio of 1H12C14N to 1H13C15N in sample; cx is the concentration of the unknown 1H12C14N in the sample in nanograms per gram or nanograms per millilitre; wx is the weight or volume of an unknown sample in grams or millilitres; cs is the concentration of spike added in nanograms per gram or nanograms per millilitre; ws is the weight of the spike added in grams or millilitres; I1x is the fraction of weight that is native (unlabelled) 1H12C14N in the sample; I1s is the fraction of weight that is native (unlabelled) in spike (assumed to be close to zero); I2x is the fraction of weight that is enriched (labelled) in sample (assumed to be close to zero); and I2s is the fraction of weight that is enriched (labelled) 1H13C15N in the spike. Consider the internal standard whose concentration of 13C15N- in 0.1 N sodium hydroxide is known. If ws g of cs ng/g internal standard is added to wx g whose concentration cx is unknown, in the true spirit of isotope dilution, if the ratio of 1H12C14N abundance to that of 1H13C15N is measured by examining a chromatogram while knowing the molecular fraction of native and labelled, cx can be calculated. The contributions from I1s and I2x as shown in equation 6 are negligible and these terms approach zero. Equation 6 can be simplified, rearranged, and solved for cx to give:[7]

Figure 3: Hypothetical plot of Rm versus cx drawn from data found in Table 3. This illustrates isotope dilution MS in its purest form.
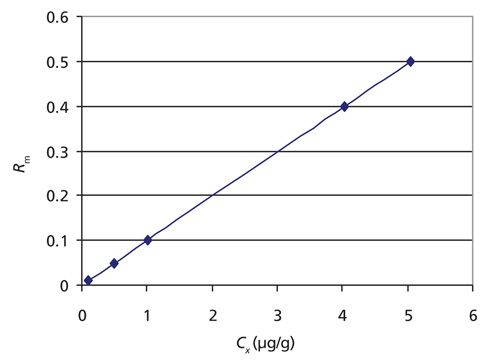
Murphy and colleagues at NIST provided numerical values for I2s and I1x. The molecular fraction of 1H12C14N is 0.98519 and that of 1H13C15N is 0.9948 (10). Table 3 gives various scenarios as cx is calculated for various hypothetical combinations of weights and concentrations with hypothetical data for Rm based on equation 7. In the true spirit of isotope dilution, if experiments 1-5 in Table 3 are plotted as Rm versus cx, a linear calibration plot is evident as shown in Figure 3. I have demonstrated that the possibility exists to quantitate CN- in whole blood samples without calibration.
A Decade of Method Development:
The sections that follow outline a decade of continuous quality improvement and refinements of the original method (9). First, I discuss changes made in reagent addition to the headspace vial. Headspace sampling changes are discussed next followed by changes in PLOT column performance. Finally, I share a specific example of isobaric interference that occurred in my laboratory.

Adding Internal Standard:
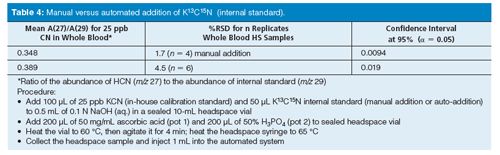
Automatic Versus Manual: It became clear some time ago that the autosampler could automatically add the internal standard (K13C15N dissolved in 0.1 N sodium hydroxide at parts-per-million levels). The internal standard must be added before the ascorbic acid and phosphoric acid reagents are added to the blood matrix sample before headspace sampling. This would eliminate manual pipetting. Table 4 shows results for replicate samples comparing manual versus automated internal standard addition to whole blood samples for the lowest calibration standard (25 ppb CN-). This early study demonstrated that automated robotic pipetting of the internal standard could replace manual pipetting without sacrificing precision and accuracy in the quantification of HCN in whole blood samples.
Replacing Separate Ascorbic Acid and Phosphoric Acid Reagent Additions with One Mixed Acids Reagent Addition While Lowering the Phosphoric Acid Strength:
Ascorbic acid is added to blood specimens before headspace sampling to minimize interference from the weak base thiocyanate (SCN- ) (10). Phosphoric acid is then added to provide the necessary hydronium ions (H3O+) to protonate CN- before headspace sampling. The original method called for ascorbic acid reagent addition from a first vial followed by phosphoric acid addition from a second vial (9). Early on, I explored the effect of prior mixing of both reagents in a single vial before the addition to the blood sample on CN- quantification. It made little difference in analytical outcomes whether strong and weak acids were added in sequence or added as previously mixed acids.
Optimizing the Acid Strength of Phosphoric Acid:

Figure 4: Bar graph showing the measured pH after stepwise reagent–sample addition using 5% phosphoric acid.
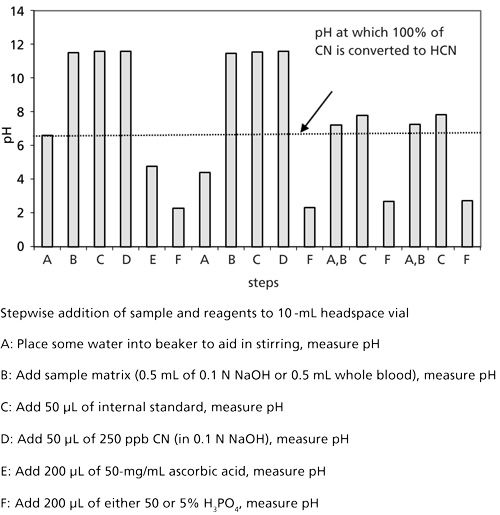
Earlier studies of the pH of the aqueous/blood matrix initially made alkaline using ~0.1 N sodium hydroxide, and measuring solution pH after each reagent addition are shown for three replicate samples in Figure 4, steps A through F (19). The pH at which CN- is completely converted to HCN, obtained from the interpretation of Figure 1, is shown as the horizontal line in Figure 4. Irrespective of the more dilute or more concentrated phosphoric acid, the pH measured after the acid has been added (step F) is well below the cutoff (horizontal line in Figure 4). An optimum acid strength of 20% phosphoric acid is common practice in many SPHLs today.

Increasing Throughput by More Creatively Programming the Autosampler Sequence:
In the original method, the first headspace vial in the sample tray received all reagents. This headspace vial was then transported to the heated agitator and subsequently headspace sampled and injected. The cycle repeats itself for each and every sample vial in the tray. The autosampler can be reprogrammed to have the upper rail syringe head mount add all reagents to all headspace vials in the tray. After all headspace vials have received reagents, each headspace vial is brought to the agitator, heated, headspace sampled, and injected. Details of programming the autosampler are found in the experimental section. Studies were conducted at the CDC and efficiencies were realized (20). A second prep method is needed if, for whatever reason, the automated sequence is stopped. An “inject only” autosampler program was developed to address this shortcoming. This prep sequence brings the headspace vial to the agitator, heats the sample and headspace samples, and injects the sample without reagents being added. It is surprising just how many times I had to resort to using the “inject only” prep sequence.
Headspace Sampling and GC Inlet Considerations:
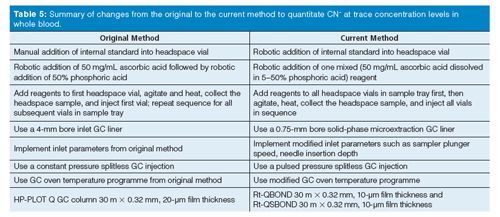
Further refinements to the original method include changes to headspace sampling plunger speed, changes to the needle insertion depth; changing from a 4-mm bore GC inlet liner to a 0.75-mm bore solid-phase microextraction (SPME) GC inlet liner, and changing from constant pressure to pulsed pressure GC injection. These refinements were presented by the CDC to SPHLs (20).

Figure 5: EICs for m/z 26, 27, 29 comparing a “more selective” H-P Plot Q column (left) and a “less selective” column (right) under identical GC conditions. HCN retention time is 4.34 min. Temperature programme: 100 °C hold for 0.0 min then to 120 °C at 4 °C/min; hold for 0.0 min,then to 250 °C at 99 °C/min, hold for 1.69 min; post temp 50 °C; max temp 270 °C; equilibration time 0.01 min.
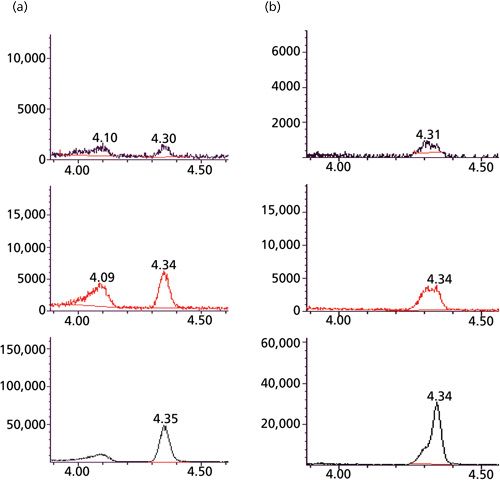
Inconsistent PLOT Column Performance:
Figure 5 show two adjacent extracted ion chromatograms (EICs) for HCN obtained from a blood sample using the automated cryotrapping isotope dilution headspace GC-MS technique. The optimum EIC on the left displays a Gaussian peak shape, a narrow peak width, and a reproducible HCN retention time. Analysts can expect coefficient of determinations (r2), and QC criteria to be met. In contrast, the nonoptimum EIC on the right displays non-Gaussian peak shape and larger peak widths. Analysts can expect r2 and QC criteria to be unmet. PLOT columns have shown an inconsistency with respect to peak shape and peak width over the past decade. The recommended PLOT column about a decade ago was the 30 m × 0.32 mm, 20-µm (film thickness) H-P Plot Q (Agilent Technologies). Over time, some laboratories experienced inconsistent PLOT column performance:
- Poor resolution between CN and propane
- Poorly shaped peaks
- Reduced intensity
- No peaks at all
Both the Rt-QBond and the Rt-QSBond columns (Restek Corp.) have been tested at the CDC as well at as several SPHLs. The Rt-QBond column has a 100% divinylbenzene stationary phase (nonpolar), and the Rt-QSBond column is divinylbenzene with a small amount of 4-vinylpyridine incorporated into the structure (intermediate polarity). I have observed that the Rt-QSBond column shows significantly greater chromatographic resolution between HCN and propane when compared to the Rt-QBond column. Improving PLOT column performance to better quantitate a fixed gas such as HCN remains an active area of pursuit. A specific limitation of the automated cryotrapping isotope dilution headspace GC-MS technique during my watch occurred a few years ago.

Figure 6: Headspace GC–EI-MS total ion chromatogram of QC low range under SIM conditions: Column: QBond (Restek). Temperature programme: 105 °C, hold for 6 min to 200 °C, 0.9 mL/min flow rate. Propane at a retention time of 4.572 min, HCN (4.653 min), and presumably methyl mercaptan (5.320 min).
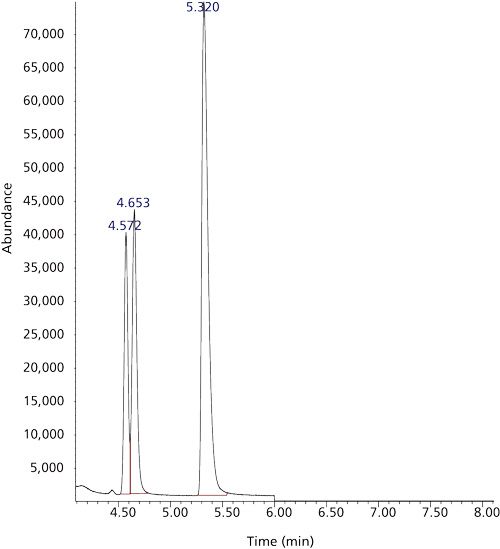
Isobaric Matrix Interference:
Figure 6 shows the isobaric interferences caused by the presence of traces of propane in the blood specimen when using the Rt-QBond PLOT column. The partially resolved dual peaks at ~4.6 min shown in the chromatogram were obtained by abandoning the temperature programme in favour of an isothermal GC run and lowering the column flow rate from 1.0 mL/min to 0.9 mL/min of helium carrier gas. This isobaric interference was first noted in the QC blood samples (stored at -20 °C). These QC blood samples were inadvertently and repeatedly brought to room temperature because of inconsistent maintenance of the cold storage temperature. When operating under the temperature programme conditions of the method, this isobaric interference significantly increased the abundance of m/z 29 since it was coeluted with HCN. The significant increase in the internal standard abundance at m/z 29 led to our reporting of a consistently biased low result for the number parts per billion HCN for all the QC blood specimens at that time.

Figure 7: Headspace GC–EI-MS TIC of propane under scan conditions. Column: QS-Bond (Restek). Temperature programme: 100 °C to 120 °C at 4 °C/min then heat rapidly to 200 °C. The peak at 3.968 is because of propane. The peak at 5.718 is unidentified.
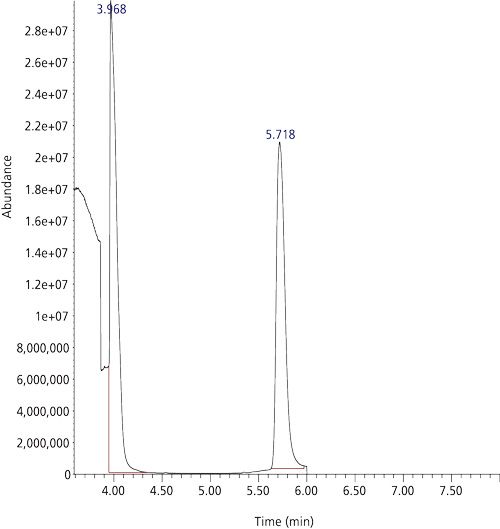
Next, I installed the Rt-QSBond PLOT column. I found that this column could chromatographically resolve HCN from propane. I returned to the oven temperature programme of the original method. A 40-mL volatile organic analysis (VOA) vial was purged, then filled with propane, and the headspace was sampled. Figure 7 shows a chromatogram (under scan conditions) for the contents of the VOA vial purged with propane. A mass spectrum at the apex of the peak at 3.968 min is shown in Figure 8. The large abundance in the mass spectrum is because of a fragmented ethyl moiety at m/z 29!

Figure 8: EI mass spectrum for propane for the TIC chromatogram in Figure 7. Evidence for the isobaric interference since propane has a base peak at m/z = 29.
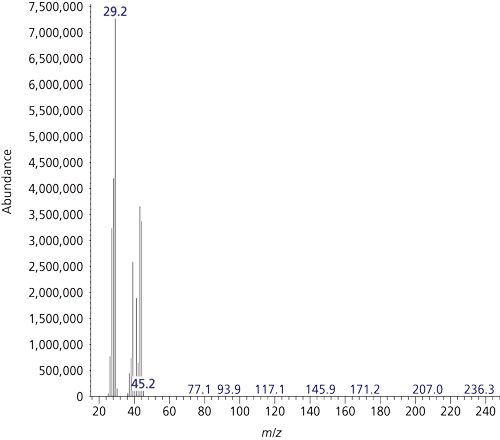

Figure 9: Headspace cryotrapping GC–EI-MS TIC: QC mid range from under SIM conditions: Column: QS-Bond (Restek). Temperature programme: 100 °C to 120 °C at 4 °C/min then heat rapidly to 200 °C. The retention time for HCN is 4.753 min. All other peaks are unidentified.
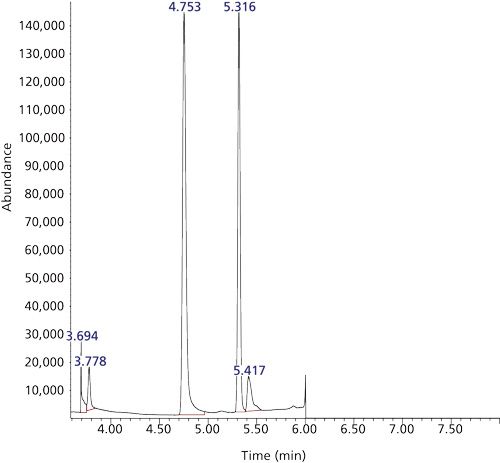
Figure 9 shows a chromatogram for a QC medium range (~140 ppb HCN) blood specimen using the Rt-QSBond column and demonstrates more than adequate chromatographic resolution between propane and HCN while exhibiting good Gaussian peak shape, low peak width at half height, and adequate theoretical plates. The isobaric interference as I observed and discussed above represents a severe limitation when using GC-MS to quantitate lowâmolecular-weight analytes such as HCN. A recent contribution has suggested that the analysis of low mass species presents an opportunity to investigate a new detector such as vacuum ultraviolet absorption (VUV) for GC (21). T
he Need for Periodic Autosampler and GC-MS Maintenance:
Periodic instrument maintenance in the quantification of HCN by automated cryotrapping isotope dilution headspace GC-MS is critical for adequate preparedness in the event that a laboratory becomes engaged in quantifying HCN in human blood due to exposure. It is recommended that tension (bungee) cords for the autosampler robotic head mount be replaced at a frequency of once per year. As the tension cords deteriorate, the tendency for a headspace vial to get hung up between the bottom of the head mount and the sample tray increases. Preventative maintenance on the GC-MS instruments should be done at least once per year. Periodic ion source cleaning is essential. Monitoring ion source filament resistance guards against filament burnout. Written logs for maintenance and continued vigilance are key tools to properly maintain this instrumentation and ultimately guarantee good laboratory practice.
Conclusion
The importance of understanding the influence of secondary equilibria (pH) and analyte gas-liquid headspace partitioning from dilute solutions (Henry’s law) is critical to identifying and quantifying CN- in aqueous and whole blood matrices. Equations from the literature were used to prove that calibration could be eliminated by applying isotope dilution in its purest sense. In 2003, I was asked to develop a rugged method to quantitate CN- in blood using a computer controlled dual rail robotic auto-sampler atop a conventional GC-MS instrument. Training was provided by the CDC yet numerous technical obstacles had to be overcome to meet stringent quality assurance (QA) and QC requirements. Over the years, advantages and limitations in the original method were identified and led to specific method improvements that used more of the robotic autosampling technology and experimental evidence justified reducing the strength of phosphoric acid and mixing ascorbic acid with phosphoric in a mixed acids reagent.
Acknowledgements
The author is grateful for the support he received from the Michigan Public Health Institute, Okemos; the Michigan Department of Community Health, Bureau of Laboratories, Lansing; and the Department of Health and Human Services, Centers for Disease Control and Prevention, Public Health Emergency Preparedness. The technical support teams from Gerstel, USA, and from Agilent Technologies, Inc. are gratefully acknowledged. The constructive suggestions from the reviewer to make this article more understandable are appreciated.
References
The Merck Index
, 13th Edition, M. O’Neil, Ed. (Merck & Co. Inc., 2001).
Mayo Medical Laboratories Interpreted Handbook
, D. Leavelle, Ed.(Mayo Clinic, 2001). R. Berry, S. Rice, and J. Ross,
Physical Chemistry
(Oxford University Press, 2000), p. 407. D. Mayo, F. Miller, and R. Hannah,
Course Notes on the Interpretation of Infrared and Raman Spectra
(Wiley-Interscience, 2004), p. 322. J. Ma and P. Dasgupta,
Anal. Chim. Acta
673
, 117-125 (2010). “Determination of Cyanide in Drinking Water by GC/MS Headspace Analysis” (ME355.01 Rev 1.0, 5/26/2009, and Gerstel Solutions Worldwide, March, 2010), pp. 12-13. C. Männel-Croisé and F. Zelder,
Inorg.Chem.
48
, 1272-1274 (2009). W.C. Blackledge, C.W. Blackledge, A. Griesel, S.B. Mahon, M. Brenner, R.B. Pilz, and G.R. Boss,
Anal. Chem.
82
, 4216-4221 (2010). A. Calafat, Rapid Quantitation of Cyanide in Whole Blood by Automated Headspace Gas Chromatography (Centers for Disease Control and Prevention [CDC], Atlanta, Georgia, Draft Method, 2001). K. Murphy, M. Schantz, T. Butler, B. Benner, Jr., L. Wood, and G. Turk,
Clin. Chem.
52
(3), 458-467 (2006) . D. Harris,
Quantitative Chemical Analysis
, 3rd Edition, (Freeman, 1991). E. Moelwyn-Hughes,
Physical Chemistry
, 2nd Edition (Pergamon Press, 1961), pp. 768-772. R. Hites,
Elements of Environmental Chemistry
(Wiley-Interscience, 2007), chap. 5. J. Ma, P. Dasgupta, W. Blackledge, and G. Boss,
Environ. Sci. Technol.
44
(8), 3028-3034 (2010). B. Kolb and L. Ettre,
Static Headspace Gas Chromatography: Theory and Practice
(Wiley-VCH, 1997), chap. 2. W. Budde,
Analytical Mass Spectrometry
(Oxford University Press, 2001), p. 84. J. Fassett,
Pure & App. Chem.
67
(11), 1943-1949 (1995), J. Fassett and P. Paulsen,
Anal. Chem.
61
, 643A-648A (1989). P. Loconto, “Toward a Leaner but not Meaner Quantitative Determination of Chemical Warfare Agents,” LRN-C, Oakland, California, USA, April 2004. M. McGuigan, “Detection of Hydrogen Cyanide in Blood Using GC-MS with Headspace Sample Injection,” ERCL-C, Salt Lake City, Utah, USA, July 2010. K. Schug and H. McNair,
LCGC North Am.
33
(1), 24-33 (2015).
Paul Loconto
recently retired from the Michigan Department of Community Health, Bureau of Laboratories, in Lansing, Michigan, USA, where he focused on developing and implementing analytical methods to quantitate trace organics from aqueous, blood, serum, and fish tissue matrices. Direct correspondence to: loconto@att.net

















Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



