Data Integrity Focus, Part IX: Understanding Data Integrity Metrics for Chromatography
LCGC North America
Here, we review the regulatory expectations of metrics for monitoring data integrity and discuss their practical implementation in a regulated chromatography laboratory.
One area of a data integrity program is the establishment of metrics for monitoring data integrity. Here, we will look at the regulatory expectations and discuss their practical implementation in a regulated chromatography laboratory.
This is the ninth article in a series on data integrity focus in the regulated chromatography laboratory (1–8), and there was also a six-part series on data integrity in 2018 co-authored by Mark Newton and myself (9–14). In two of these articles, a Data Integrity Model was presented (1,10) that consisted of a foundation layer and three levels above. One of the key items in the foundation layer is management leadership for data integrity, and this is reflected in both EU GMP Chapter 1 regulations (15), as well as data integrity guidance documents from regulatory authorities, including the Food and Drug Administration (FDA) (16), the Medicines and Healthcare Products Regulatory Agency (MHRA) (17), the World Health Organization (WHO) (18), the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S) (19), and industry guides (20,21).
These guidance documents set a regulatory expectation that management and companies should be able to detect data errors and falsification via either second person review or quality oversight, such as data integrity audits and investigations. The alternative is a starring role in warning letters posted on the FDA's wall of shame with citations such as:
- There was a lack of basic oversight by operations, quality unit, and site managers, as rewriting and destruction of original current good manufacturing practices (CGMP) records was allowed to persist over a significant period without implementation of systems and controls to prevent data manipulation (22).
- Your senior management, at the local and corporate levels, is responsible for ensuring that strict corporate standards, procedures, resources, and communication processes are in place to detect and prevent breaches in data integrity, and that such significant issues are identified, escalated, and addressed in a timely manner (23).
As can be seen, management held accountable for lapses in data integrity of their staff. The FDA is also interested in quality metrics, as we shall now see.
Quality Metrics and Data Trending
From a laboratory perspective, knowing the out-of-specification (OOS) rate is an important criterion for both quality and data integrity. One of the key questions to ask when auditing or inspecting a laboratory is to ask to see the OOS results for the past six months. However, answering, "we don't have any" can result in a regulatory surprise such as:
Since beginning manufacturing operations in 2003, your firm has initiated a total of 0 out of specification investigations for finished product (24).
Even when you have OOS results, but the overwhelming majority of them are invalidated, that can result in a winning entry for an FDA warning letter:
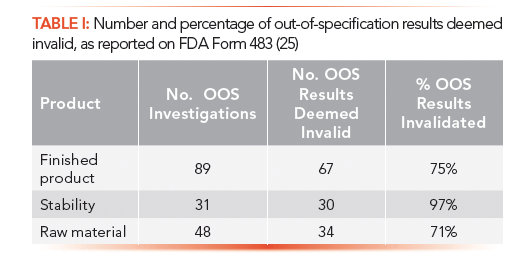
Observation 1: There is a failure to thoroughly review any unexplained discrepancy and the failure of a batch or any of its components to meet any of its specifications whether or not the batch has been already distributed. Specifically, from January 2016 to March 2017, your firm invalidated several OOS results as summarized in Table 1 (25).


Given that analytical procedures are subject to variation, we expect to see not only OOS, but also out-of-expectation (OOE) and out-of-trend (OOT) results. There is a specific EU GMP regulation that requires trending of laboratory results:
6.9 Some kinds of data (e.g. tests results, yields, environmental controls) should be recorded in a manner permitting trend evaluation. Any out of trend or out of specification data should be addressed and subject to investigation (26).
6.16 The results obtained should be recorded. Results of parameters identified as quality attribute or as critical should be trended and checked to make sure that they are consistent with each other.... (26).
The FDA has focused on quality metrics as a way of identifying lower risk facilities, and has issued two draft guidance for industry documents on quality metrics for comment in July 2015 and November 2016 (27). To monitor the performance of quality control (QC) laboratories, the agency has selected the invalidated out-of-specification rate (IOOSR), which is defined as the number of OOS test results for lot release and long-term stability testing invalidated as a percentage of total OOS results. Seeing the results in Table I, you can understand why the FDA has selected this metric.
What is the OOS rate in your laboratory? If you don't know, there might be some issues during the next inspection.
Quality and Data Integrity Metrics
Tracking quality or data integrity metrics may sound like yet another task for chromatographers to perform in addition to their normal work, but many of you already are used to working with them under the name of key performance indicators (KPIs). KPIs are used by many laboratories to measure the operational and quality performance of processes.
For example, laboratory managers may need to know how many samples are being analyzed, and how long it takes to analyze them. They need to know if there are any bottlenecks, and if they have enough resources assigned to handle the work. Metrics can provide the information to measure a process or activity. If used correctly, metrics help staff understand the performance measures that a laboratory is judged against. Some common laboratory metrics include numbers of samples analyzed by unit time, turnaround time (TAT) per sample measured against targets, instrument utilization rate, and numbers of injections per chromatograph per unit time. The same approach can be taken with metrics to assess data integrity within a laboratory or organization. I can see you roll your eyes: more work, you think!
Metrics Must Be Generated Automatically
A key requirement is that metrics must be collected automatically. You may ask why. The simple reason is that if humans collect and collate the data manually, it is an error-prone process, tedious, labor intensive, and could also be subject to falsification. The latter is not the best option when generating data integrity metrics.
Automatic metric generation is the only way to create metrics that are timely, accurate, and repeatable. This is where suppliers of chromatography data systems (CDS) and other laboratory informatics applications could be more helpful by implementing applications that generate both general and data integrity metrics automatically.
Why Metrics for Data Integrity?
Why are data integrity metrics important? It is now a regulatory expectation, as we can see from the excerpt of PIC/S guidance PI-041 shown in Table II (19). There is an expectation that data integrity metrics of processes and systems are collected for management review and that management reviews them, and acts if there are any issues.


Could Metrics Influence Behavior?
Look at the second paragraph in section 6.5.1 in Table II that says to take care when selecting a quality metric that could result in a culture in which data integrity is lower in priority. Let us consider this statement further. Take the example of sample TAT. Imagine that the laboratory target for TAT is set for 10 days, and over the past few months the target has been missed and the laboratory manager is very peeved (this is understandable if he or she gets a bonus based on laboratory performance). The sample you are starting to analyze has been in the laboratory for nine days, and your analysis will take two days to perform. You will miss the TAT target time, unless...
This is where quality culture, management, and data integrity collide, and it is where the PIC/S guidance caution about metric comes into play. Can some metrics, intended to monitor activities, become the means of inducing behaviors that compromise data integrity? Possibly, in the case where a manager is putting pressure on staff to meet performance targets. Another factor to consider is that the areas that are monitored tend to improve because they get attention, but this can be at the expense of other activities that are not monitored.
Metrics for Laboratory Data Integrity
Most data integrity reports can only identify areas for further investigation, because it is rare where a single record equates to a data integrity breach (11,28). You must carefully select what areas will be monitored by metrics, because too many can bury you in data that are intended to help you. Start small, evaluate the data, and adjust as necessary. I like a proof of concept or prototyping approach: Identify a metric, generate the data, and then assess what is happening. This is especially so when reports can be collected retrospectively so that the situation both before and after the metric was established can be evaluated.
It is virtually impossible to automatically generate and analyze metrics for manual or hybrid activities. For example, you could possibly detect missing files in a system by comparing the instrument use log (paper) against a report of sample identities from all runs stored on a CDS if a falsifier was stupid enough to complete the log honestly. However, this approach is too tedious to be done on a regular basis, and is best left for either a data integrity audit or investigation as discussed in Part VI of this series (6). In contrast, sample identities in a laboratory information management system (LIMS) or electronic laboratory notebook (ELN) could be compared against the data files on laboratory data systems using an automated script. Owing to the time involved in their preparation, manual reports should be limited to special situations, such as detecting fraud from a single suspected individual as part of an investigation.
Some data integrity issues can't be monitored or checked, such as ensuring the correct sample volumes or weights are taken during manual preparation prior to analysis. Data recorded manually outside of a computerized system are very difficult to verify but this is where data acquisition to a LIMS or ELN can improve the situation compared with the following citation:
He said that he recalibrated the balance and prepared new documentation, and subsequently discarded the original record. Furthermore, we learned that additional original calibration records of other balances had similarly been discarded (29).
Metrics could also include assessment of the performance of individuals, such as measuring the time to complete a method for several different people and looking for someone working too quickly. This is where regulatory compliance clashes with, in some countries, the works council or labor authorities who are concerned with protecting the rights of workers. The balance between ensuring data integrity of the organization producing and marketing pharmaceutical products needs to be balanced with the rights of individuals. However, the regulatory expectation is that processes must ensure the integrity of records generated.
There are some activities that are just difficult to catch with a report. For example, once a user changes the naming convention on repeat injections, you will probably not detect it on a routine report. And trying to catch everything can only be done if you have unlimited resources. So, what can we do practically?
Some Suggested Metrics for Chromatography
Some suggested data integrity metrics for chromatography processes are presented below and include:
- Aborted runs: Reporting runs that were started, but not concluded, can point toward analysts looking at data during the run, then making the decision to terminate the run to avoid accepting data they believe may be out of specification or trend. Aborted runs, in a well-controlled GMP environment, should always be viewed with a suspicious eye.
- Short runs: Reporting runs that have fewer than an accepted number of injections (for example, 1–3 injections) is a means of detecting analysts who re-inject a sample to obtain a new result that can replace one from a previous injection or who want to see what the result could be.
- Run evaluation sequence: As mentioned by Newton and McDowall (9), there should be a procedural order for processing a chromatography run: (1) evaluation of system suitability; (2) evaluation of reference standard acceptability; (3) evaluation of method acceptance criteria; (4) evaluation of sample results.
- It is possible to create reports that ensure this sequence of events is happening, based on time stamps of events. This report can point toward analysts evaluating sample results before other acceptance criteria, then finding means to reject the run, such as manipulating standards or suitability to ensure failure of the run, a type of "testing into compliance."
- Recalculated dataset: Monitoring runs that are calculated more than once has two benefits: It is one means of looking across runs for potential improper activities, but it also can point out methods that are not well configured, and therefore require additional manual intervention. Recalculations and manual integrations not only have data integrity impact, but lab efficiency as well.
- Manual integration: For each analytical method at each site, report the number of peaks automatically integrated and manually integrated. This metric provides insights that lead to more automated integration. For example, site A automatically integrates 80% of all peaks for method Y, while all other sites using the same method automatically integrate only 30% of their peaks. What do analysts at site A know about this method that permits such a high level of automated integration? What can the rest of the organization learn from site A?
- Organizational benchmarking: For each report type, generate a summary report that compares the number of records found by site. This permits comparisons, so that managers can look for sites that have unusually high (or low) activity compared to other sites. For example, a site with twice the number of aborted runs as other sites might lead to a QA inquiry to understand the high number of aborts; perhaps equipment issues, a fragile method, or poor behaviors are the root of the issue, but the report creates the signal that starts the investigation.
Learning from Others' Mistakes
One of the best ideas for monitoring metrics often come from regulatory enforcement actions such as FDA warning letters. The key requirement is to read the cited deficiency and ask yourself, "How would we detect this situation in our own laboratory (28)?" This question will cause you to think about the data patterns that accompany the behavior, and then to formulate a query that could detect the data pattern. For example, a firm is cited for manipulating the system clock to falsify timestamps in audit trail records. If this happens, there could be a series of system audit trail entries, one for each clock adjustment. In addition, there will be some frequently written audit trails (such as inter-system messages) where the clock will appear to go backward because of the clock manipulation. So a query that checks for clock entries that do not continue to increase could flag the clock manipulation behavior. Just be careful when there are summer and winter time changes.
Limitations of Metrics
It is important to remember that all metrics are not created equal; some will prove more effective than others in your operation. In addition, metrics seldom identify a true issue with every reported record in a report; rather, they highlight suspicious records that require a human to investigate; this requires a time investment, and therefore becomes a limitation on reporting effectiveness. Finally, some real issues will not be detected in a report, such as reanalyzing a sample on a simple instrument (for example, a pH meter), or picking the desired outcome and forwarding it to LIMS: This is a data integrity issue that will not be detected on any report.
Summary
Tracking metrics for data integrity is a regulatory expectation. This article has provided an overview and background about quality metrics and which of those to collect for a chromatography laboratory. Metrics should be generated automatically and be used to detect trends that require human investigation. Please avoid paralysis by analysis by having too many metrics and the accompanying data: Start small and work from there.
References
(1) R.D. McDowall, LCGC N.Amer. 37(1), 44–51 (2019).
(2) R.D. McDowall, LCGC N.Amer.37(2), 118–123 (2019).
(3) R.D. McDowall, LCGC N.Amer.37(3), 180-184 (2019).
(4) R.D. McDowall, LCGC N.Amer.37(4), 265–268 (2019).
(5) R.D. McDowall, LCGC N.Amer. 37(5), 312–316 (2019).
(6) R.D. McDowall, LCGC N.Amer. 37(6), 392–398 (2019).
(7) R.D. McDowall, LCGC N.Amer. 37(8), 532–537 (2019).
(8) R.D. McDowall, LCGC N.Amer. 37(9), 684–688 (2019).
(9) M.E. Newton and R.D. McDowall, LCGC N.Amer.36(5), 330–335 (2018).
(10) M.E. Newton and R.D. McDowall, LCGC N.Amer.36(1), 46–51 (2018).
(11) M.E. Newton and R.D. McDowall, LCGC N.Amer.36(4), 270-274 (2018).
(12) M.E. Newton and R.D. McDowall, LCGC N.Amer.36(7), 458–462 (2018).
(13) M.E. Newton and R.D. McDowall, LCGC N.Amer.36(8), 527–529 (2018).
(14) M.E. Newton and R.D. McDowall, LCGC N.Amer.36(9), 686–692 (2018).
(15) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 1 Pharmaceutical Quality System. 2013, European Commission: Brussels.
(16) FDA Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers (Food and Drug Administration, Silver Spring, Maryland, 2018).
(17) MHRA GXP Data Integrity Guidance and Definitions (Medicines and Healthcare Products Regulatory Agency, London, United Kingdom, 2018).
(18) WHO Technical Report Series No. 996 Annex 5 Guidance on Good Data and Records Management Practices (World Health Organization, Geneva, Switzerland, 2016)
(19) PIC/S PI-041-3 Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments Draft (Pharmaceutical Inspection Cooperation Scheme, Geneva, Switzerland, 2018).
(20) GAMP Guide Records and Data integrity (International Society for Pharmaceutical Engineering, Tampa, Florida, 2018).
(21) GAMP Good Practice Guide: Data Integrity-Key Concepts (International Society for Pharmaceutical Engineering, Tampa, Florida, 2018).
(22) FDA Warning Letter Sun Pharmaceuticals (Food and Drug Administration, Rockville, Maryland, 2014).
(23) FDA Warning Letter Fresenius Kabi Oncology (WL: 320-13-20) (Food and Drug Administration, Rockville, Maryland, 2013).
(24) FDA 483 Observations, Sri Krishna Pharmaceuticals Limited (Food and Drug Administration, Silver Spring, Maryland, 2014).
(25) FDA 483 Observations: Lupin Limited (Food and Drug Administration, Rockville, Maryland, 2017).
(26) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 6 Quality Control (European Commission, Brussels, Belgium, 2014).
(27) FDA Guidance for Industry Submission of Quality Metrics Data, Revision 1 (Food and Drug Administration, Rockville, Maryland, 2016).
(28) M.E. Newton and R.D. McDowall, LCGC Europe30(12), 679–685 (2017).
(29) FDA Warning Letter Leiner Health Laboratories (Food and Drug Administration, Rockville, Maryland, 2016).
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com






: An Interview With Fabrice Gritti")




, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.


Using GC-MS to Measure Improvement Efforts to TNT-Contaminated Soil
April 29th 2025Researchers developing a plant microbial consortium that can repair in-situ high concentration TNT (1434 mg/kg) contaminated soil, as well as overcome the limitations of previous studies that only focused on simulated pollution, used untargeted metabolone gas chromatography-mass spectrometry (GC-MS) to measure their success.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

Potential Obstacles in Chromatographic Analyses Distinguishing Marijuana from Hemp
April 28th 2025LCGC International's April series for National Cannabis Awareness Month concludes with a discussion with Walter B. Wilson from the National Institute of Standard and Technology’s (NIST’s) Chemical Sciences Division regarding recent research his team conducted investigating chromatographic interferences that can potentially inflate the levels of Δ9-THC in Cannabis sativa plant samples, and possible solutions to avoid this problem.

