Advancing Gene Therapy: Enzyme Selection for Effective RNA Oligonucleotide Mapping
New gene therapy modalities, such as CRISPR guide RNA (single guide ribonucleic acid [sgRNA]) and messenger RNA (mRNA), continue to make progress in both primate and first-in-human trials. As this progress builds, the industry remains accountable for characterizing these molecules to meet the requirements of regulatory authorities. The sequence of ribonucleic acid chains is a critical quality attribute (CQA) as it significantly influences the drug’s efficacy. Moreover, in the case of CRISPR molecules, any ambiguity in sequence can also translate to undesired off-target effects. Similarly to peptide mapping in therapeutic protein characterization, mRNA oligonucleotide mapping by mass spectrometry (MS) is the preferred method because of its ease of implementation in regulated environments when compared to next-generation sequencing (NGS). One of the aims of this study was to make oligonucleotide mapping as accessible as peptide mapping. Oligonucleotide mapping gives a direct molecular compositional analysis of drug substance components. It also has the potential to support multi-attribute monitoring (MAM) approaches, such as 5’-capping determination and 3’ poly(A) tail length distribution in mRNA molecules, as well as the pinpoint confirmation of important modifications incorporated into CRISPR guides. In this study, four enzymes were evaluated—RNase T1, RNase 4, Cusativin, and MC1 —for oligo mapping of sgRNA and mRNA of varying lengths. The impact of missed cleavages on sequence coverage as determined by bioinformatics tools will be highlighted, as well as the effect of shorter oligos on sequence coverage.
In recent years, new therapies have been developed that rely on oligonucleotide sequences such as single guide ribonucleic acid (sgRNA) or messenger RNA (mRNA). sgRNA, which are approximately 100 nucleotides long, guide the Cas9 endonuclease to perform CRISPR gene editing at specific targets on genomic DNA (1). Meanwhile, mRNA, with sequence lengths ranging from a few hundred nucleotides to more than 10,000 nucleotides, are mostly used in the development of prophylactic vaccines, targeted protein expression, and gene replacement therapies (2). In addition, individualized neoantigen therapies, which are based on mRNA, hold significant promise to help cancer patients boost their body’s own ability to fight back against malignant cells (3).
Many critical quality attributes (CQA) have been defined by regulatory authorities, including the sequence characterization of nucleotide-based compounds (4). To date, recommendations for the study of sgRNA and mRNA sequences have involved high-throughput sequencing or Sanger sequencing (5). However, these methods may be too complex for GMP-compliant environments as they are regulated by a high number of guidelines and often require bioinformatic expertise (6). A further drawback of these approaches is their limitations when modified nucleotides are used to decrease the immunogenicity or enhance the stability of these molecules (7).
Mass spectrometry (MS) is the gold standard technique for protein sequencing through bottom-up approaches (8). It involves the digestion of proteins into peptides using selective digestion enzymes such as trypsin, followed by liquid chromatography coupled to mass spectrometry (LC–MS) analysis to confirm the sequence. Data processing reconstructs the sequence from the assigned peptide LC–MS peaks and calculates sequence coverage, indicating the percentage of the theoretical sequence that has been confirmed. Similar approaches have been adapted for sgRNA and mRNA sequence characterization, requiring enzymes to digest the long sequences into analyzable subcomponents, called oligonucleotide digestion components (7,9). Several RNA digestion enzymes with different cleavage specificity are commercially available (9–12). In this study, four different enzymes were tested: RNase T1, RNase 4, Cusativin, and MC1. These enzymes have different cleavage specificities and activities and generate unique digestion profiles. The four enzymes were evaluated using a 100-nt nucleotide sgRNA (PSMD7) molecule and three mRNA molecules of various lengths: EPO mRNA (~860 nt), Fluc mRNA (~1920 nt), and Cas9 mRNA (~4520 nt). Other specific mRNA attributes were also examined to assess the potential for multi-at-tribute monitoring (MAM), including 5’-cap characterization and poly(A) tail length determination.
Materials and Methods
Chemicals and Reagents
Acetonitrile was purchased from Biosolve. RNAse-free water and RNase T1 were purchased from Thermo Scientific. RNase 4 and NEBuffer r1.1 were purchased from NEB. RapiZyme Cusativin, RapiZyme MC1, and IonHance hexafluoroisopropanol (HFIP) were provided by Waters Technologies Corporation. Ammonium acetate and N,N-Diisopropylethylamine (DIPEA) were purchased from Sigma Aldrich. Cas9 mRNA, Fluc mRNA, and EPO mRNA were provided to Quality Assistance by a collaborator. PSMD7 sgRNA was produced by Kaneka Eurogentec for Quality Assistance in a collaborative project (PIT ATMP – Convention 8880) funded by the Region Wallonne.
Digestion of sgRNA and mRNA Enzymes
A 6.8 pmol measure of either sgRNA or mRNA was used for all digestions. Samples were diluted with ammonium acetate 200 mM pH 9 for RapiZyme Cusativin and ammonium acetate 200 mM pH 8 for RapiZyme MC1 to a final concentration of approximately 0.34 μM, and then heated for 5 min at 90 °C. After 10 min at 4 °C, 1.5 μL of reconstituted enzymes at 100 U/μL were added to the sample, followed by an incubation at 30 °C for 30 min. The enzymes were then heat inactivated by heating samples for 15 min at 70 °C.
For the digestion with RNase 4, samples were diluted with RNase-free water and heated for 5 min at 90 °C. After 10 min at 4 °C, 2 μL of NEBuffer r1.1 and 1 μL of RNase 4 (50 U) were added to the samples and incubated for 30 min at 37 °C. Finally, digestions with RNase T1 were performed after dilution in RNase-free water and incubation for 5 min at 90 °C. After cooling at 4 °C, 7.5 U of RNase T1 was added to the sample followed by an incubation of 10 min at 37 °C.
All the samples were transferred in polypropylene vials prior to injection.
LC–MS Analysis of sgRNA and mRNA Digest Products
Samples (equivalent to 5 pmol of initial sample) were loaded on a Waters Acquity Premier Oligonucleotide BEH C18 column (100 × 2.1 mm, 1.7-μm – 300 Å pore size) and eluted using a 40 min gradient from 3% to 22% of solvent B (Solvent A: 0.1% DIPEA, 1% HFIP in water–Solvent B:0.0375% DIPEA, 0.075% HFIP in 35% H2O, and 65% acetonitrile) at 0.4 mL/min, 70 °C, on a Waters Acquity UPLC H-Class Bio LC system. The Waters Xevo G2-XS QTof MS system was coupled online with the UPLC inlet and was operated in MSE negative mode. Source parameters were set as follows: capillary voltage: 1.5 kV; cone voltage: 55 V; cone gas flow: 50 L/h; desolvation gas flow: 650 L/h; source temperature: 100 °C; desolvation gas temperature: 500 °C. Mass range was set to 300–5000 m/z, scan time to 1 s, and fragmentation energy (high energy ramp) from 10.00 to 45.00 eV. The data were acquired using the compliance-ready waters_connect Informatics Platform and processed using several workflow-specific applications integrated within the platform.
Data Processing
Data processing was performed with Waters mRNA Cleaver (v1.1.0) MicroApp for in silico digestion product database generation. Oligo assignments to LC–MS peaks were performed using the waters_connect MAP Sequence App (v 1.0), and the visualization of sequence coverage with the Coverage Viewer MicroApp (v 2.0). Data independent fragmentation data (MSE) were processed by generating an in silico fragmentation list within the waters_connect SYNTHETIC Library App and assignment of fragmentation patterns with waters_connect CONFIRM Sequence App. Data were also exported and further processed inside a spreadsheet for generation of figures.
Results
Characterization of sgRNA Sequence Using LC–MS
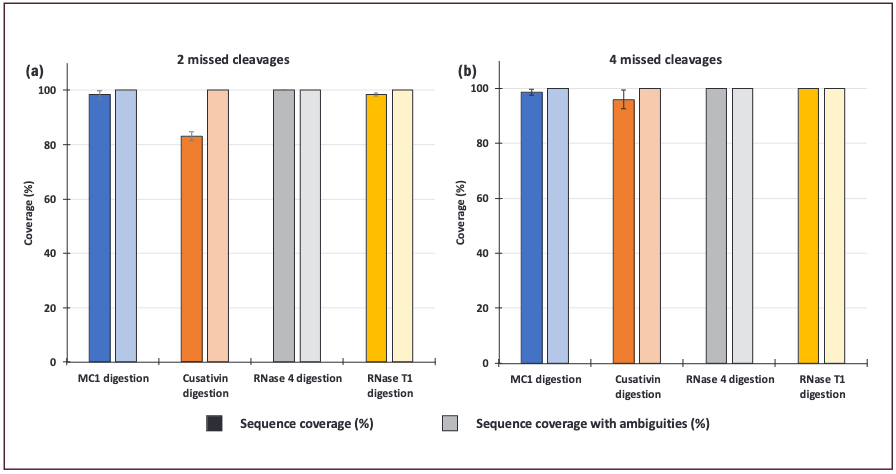
The four digestion enzymes were first tested on a 100-nt sgRNA molecule. Complete sequence coverage was only obtained with RNase 4 (with 2 missed cleavages allowed), while Cusativin led to 83% of sequence coverage (Figure 1a–dark bars) under these search conditions, and MC1 and RNase T1 98%. Those coverage values were computed based on unique oligonucleotides identified. Uniqueness was defined by the presence of a mass only existing once in the in silico digested sgRNA oligonucleotide sequence list. Ambiguous matches were defined as any case where an assignment could be made to more than one digestion component. When all potential oligonucleotide assignments were used, sequence coverage values obtained for all the enzymes were 100% (Figure 1a–light bars). However, increasing the number of missed cleavages to four led to an increase of the sequence coverage for Cusativin to 96% and for the other enzymes to 100% (Figure 1b). As this percentage was only obtained using unique oligonucleotides, it is therefore more interesting to work with longer oligonucleotide digestion components and a higher number of missed cleavages compared with the inclusion of smaller species that require structural confirmation using fragmentation data (MS/MS, MSE).

 Data obtained with 5 pmol loaded on the column and processed with 20 ppm mass tolerance and 2 missed cleavages during the oligo map search vs. (b) 4 missed cleavages. The light bars correspond to the same data as the dark ones; however, sequence coverages were also calculated taking into consideration oligonucleotides with sequence ambiguities.")
Figure 1: Sequence coverages of PSMD7 sgRNA. (a) Data obtained with 5 pmol loaded on the column and processed with 20 ppm mass tolerance and 2 missed cleavages during the oligo map search vs. (b) 4 missed cleavages. The light bars correspond to the same data as the dark ones; however, sequence coverages were also calculated taking into consideration oligonucleotides with sequence ambiguities.
Analysis of Various mRNA: The Impact of Missed Cleavages on the Sequence Coverage
As mRNA are longer species than sgRNA, digestion condition optimization was performed to obtain oligonucleotides with optimal lengths. This optimization step, performed for the four enzymes, showed that the shorter incubation time and the lower enzyme concentration were leading to the higher sequence coverage values (data not shown).
The best digestion conditions were applied to digest the three mRNA molecules. Data obtained using two missed cleavages for the process are presented in Figure 2a. Large differences in sequence coverage values obtained for the different enzymes using only unique digestion products were observed for the Cas9 mRNA, with sequence coverage at 33% for RNase T1, 47% for Cusativin, 60% for MC1, and 76% for RNase 4.

Figure 2: Sequence coverages of Fluc, EPO, and Cas9 mRNA. (a) Data obtained with 5 pmol loaded on the column and processed with 20 ppm mass tolerance and 2 missed cleavages during the search vs. (b) 4 missed cleavages.
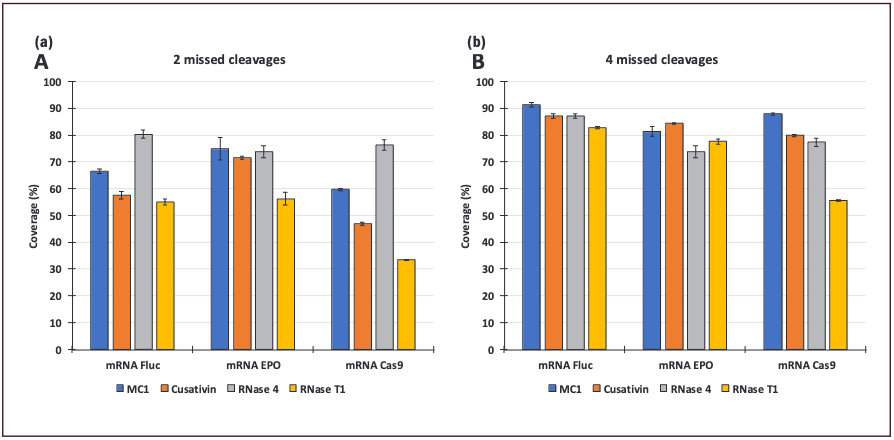
However, increasing the number of missed cleavages from two to four significantly increased the observed sequence coverages, especially for Fluc mRNA and Cas9 mRNA (Figure 2b). Coverages from RNase 4 barely increased with a higher value of missed cleavages, indicating that the digestion proceeded to a similar endpoint, no matter the adjustments made in the protocols investigated. For the Fluc mRNA, MC1 exhibited the best results with a 91% sequence coverage, while both Cusativin and RNase 4 produced coverages of 87%. RNase T1 exhibited 83% of coverage. Sequence coverage for EPO mRNA was lower and Cusativin performed the best with 84% coverage. MC1 gave the next most complete coverage (81%). Regarding the digestion of Cas9 mRNA, MC1 reached 88% of sequence coverage while Cusativin yielded 80%, RNase 4 78%, and RNase T1 56%.
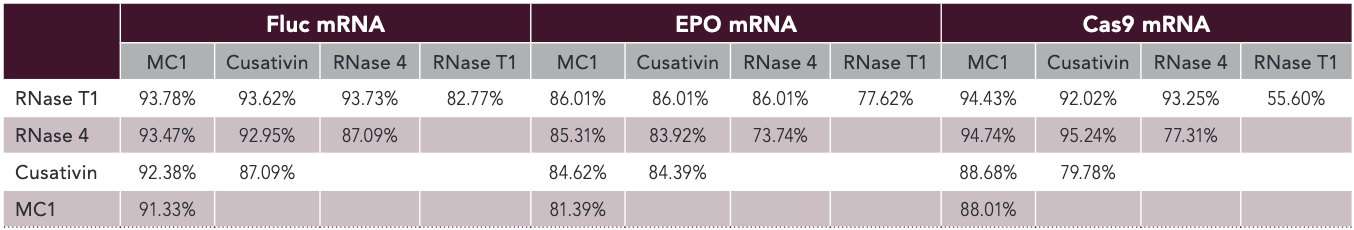
The sequence of the mRNA studied clearly influences the mapping efficiency of each RNase, suggesting that the optimal digestion conditions should be established for each molecule and enzyme combination. Alternatively, analysts could predict enzyme applicability through in silico prediction tools. The sequence coverages obtained with each enzyme are individually valuable. More interesting is the combination of results to increase total sequence coverage values (Table I). The combination of Cusativin and RNase 4 led to 95.2% of Cas9 mRNA sequence coverage compared with 80% and 78% when processed independently. An increase of approximately 2% to 6% of sequence coverage was observed for Fluc mRNA with the different combinations and 3% to 9% for EPO mRNA. However, no combination outperformed the others, and optimization should still be performed depending on the mRNA sequence studied.

Table I: Sequence coverages of the three mRNA using results from two enzymes digestion. Injections were processed independently and the sequence coverage was calculated using results from both enzymes.
Influence of Short Oligonucleotides on Sequence Coverage
The limited number of possible nucleotide mass combinations means that shorter oligonucleotides obtained from digestions often exhibit ambiguities regarding either the linear order (isomerization) of their nucleotides differing between two predicted digest products, or a single digest product appearing in multiple positions in the mRNA sequence. While the former ambiguity can be resolved by analyzing fragmentation of the digested oligos in the mass spectrometer (MS/MS or MSE) and assigning the fragmentation patterns to the linear sequence, the latter remains unsolved.
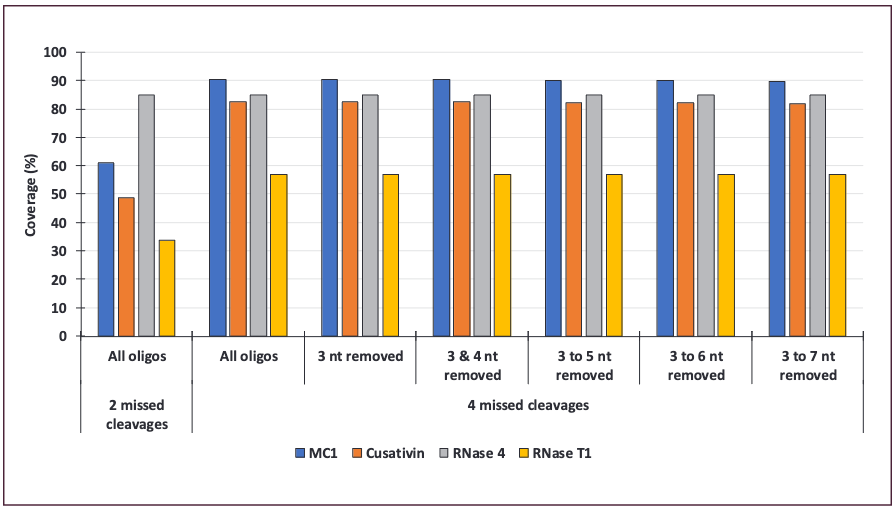
The impact of these short oligonucleotides was examined by excluding them from the sequence coverage calculations for the four enzymes (Figure 3a). Surprisingly, removing short oligonucleotides up to 4 nucleotides long had no effect on any of the coverage. Only a minimal effect was observed when excluding oligonucleotides digestion components up to 7 nucleotides long, which resulted in less than a 1% decrease in sequence coverage. Given the absence of influence of short oligos on sequence coverage, filtering steps can be added when generating the theoretical search library to reduce its size, avoid false-positive assignments, and hasten the data review process.

Figure 3: Influence of short oligonucleotides on sequence coverage of Cas9 mRNA. Data obtained after a processing step with 4 missed cleavages and 20 ppm mass tolerance and manually cured to remove oligonucleotides shorter than 3, 4, 5, 6, and 7 nucleotides with sequence coverages recalculated based on the new data.
Towards a MAM Method
Multi-attribute monitoring methods are valuable as they enable the characterization of various attributes in a single analysis. Developing fast, innovative techniques that need only small sample amounts is essential. These approaches not only conserve materials but also reduce the time needed on mass spectrometers, leading to significant operational efficiencies and cost savings. As a result of the cleavage specificity of the different enzymes used, two other critical quality attributes of mRNA could be assessed within the same data used for mRNA oligo mapping—5’-capping and 3’-poly(A) tail modifications—which are essential for mRNA translation and RNA stability upon dosing.
The intrinsic nature of the 5’-cap with its three phosphate groups prevented it from being digested by the endoribonucleases used in this study. Therefore, capping species were identified as modifications of the 5’ terminal oligonucleotide and directly assigned during the oligo map data processing workflow.
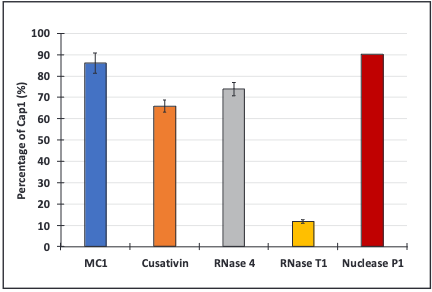
The expected cap species for Cas9 mRNA, Cap1, as well as all the potential impurities, were searched for and the results were compared with data obtained with Nuclease P1 digestion (Figure 4) (13). Overall, Cap1-capped species identified and quantified within the digest of the different enzymes were below the 90.2% observed with Nuclease P1. MC1 results were the closest to the orthogonal method, with 86% for Cap1, 65.8% for Cusativin, and 73.8% for RNase 4. RNase T1 results exhibited only 11.8% for Cap1, which was far from the expected value. This can be explained by the high number of oligonucleotides exhibiting the same masses as those containing cap impurities. Processing of fragmentation data was therefore essential to conclude the 5’-capping efficiency using RNase T1.

Figure 4: Identification and relative quantification of 5’-cap Cap1 in Cas9 mRNA sequencing analyses. Relative quantification of 5’-cap Cap1 species assigned following independent digestion with the four enzymes and orthogonal method involving Nuclease P1 digestion.
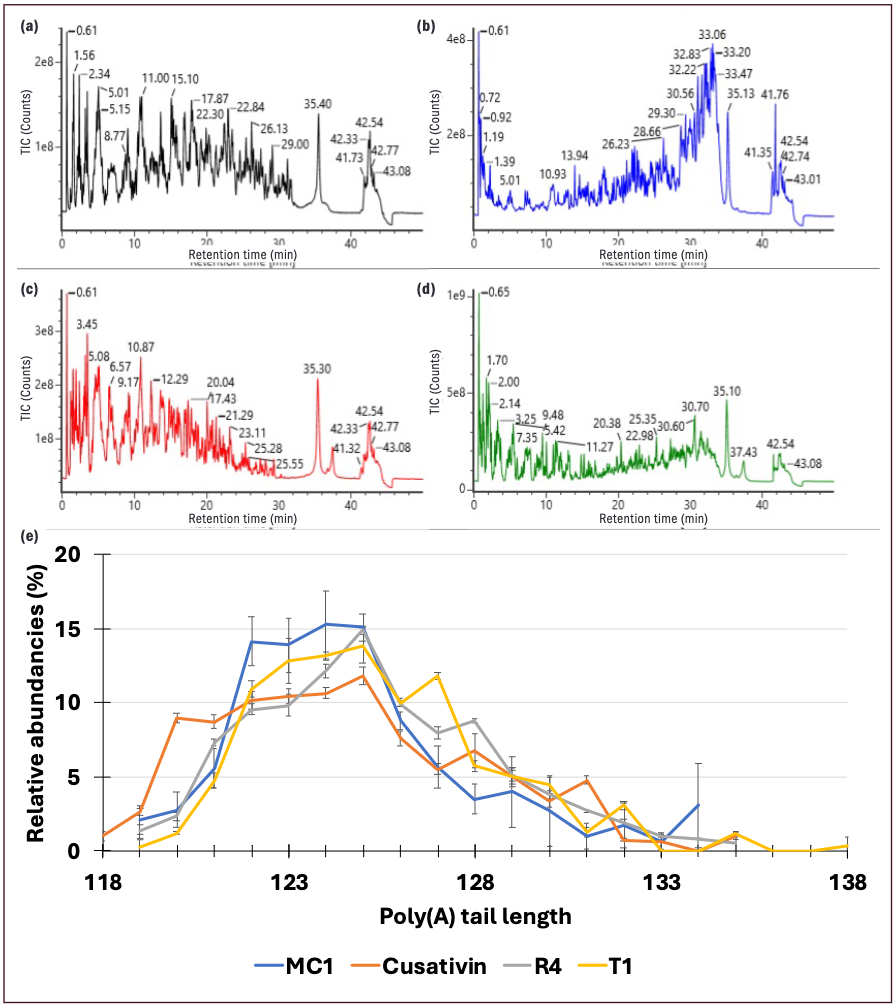
Since none of the enzymes used exhibited any activity between A nucleotides, the poly(A) tail (with additional 3’terminal mRNA coding nucleotides, depending on the cleavage specificity of the enzymes) was released following mRNA digestion. Potentially containing missed cleavages, these oligonucleotides eluted at the end of the LC gradient and, given their extended lengths, were best processed using a spectral deconvolution data processing approach. As a result of their longer sequences, it is more common to assess the distribution of poly(A) tail species in terms of the deconvoluted neutral average mass. This can be done using the same data set used for mapping but requires a separate data processing workflow to facilitate the deconvolution and assignment of poly(A) oligo dispersity. This approach was applied to Cas9 mRNA and a peak eluting at around 35 min for all the digestion conditions was observed (Figures 5a–d). From these data, the poly(A) tail species could be readily identified. Similar distributions were observed across the different enzymes used (Figure 5e), with a distribution of lengths confirmed to be present, ranging from 118 to 138 adenosine nucleotides.

Figure 5: Observation and identification of poly(A) tail attribute in Cas9 mRNA sequencing analyses. Total ion chromatogram of Cas9 mRNA digest obtained with (a) RapiZyme MC1, (b) RapiZyme Cusativin, (c) RNase 4, and (d) RNase T1. (e) Distribution of poly(A) tail lengths obtained from independent digestions with the four enzymes.
Conclusion
Oligonucleotide mapping of sgRNA and mRNA molecules by LC–MS can achieve high sequence coverage under optimized conditions. However, selecting the appropriate enzyme for digesting sgRNA or mRNA into oligonucleotides is more complex than for peptide mapping as it depends more on the specific sequence of the molecule being studied. Across the different molecules tested, different enzymes yielded optimal sequence coverage. Interestingly, it was beneficial to limit the extent of digestion by reducing enzyme quantity and incubation times. This resulted in partial digestions and a higher number of missed cleavages, which proved to be beneficial for obtaining better sequence coverage. Cusativin and MC1 were the most amenable enzymes to being tuned in this manner. This intentional under-digestion helped to reduce the abundance of short oligonucleotides that lack mass uniqueness, which can be challenging to take advantage of when characterizing longer RNA sequences. Another advantage of this approach is the ability to characterize two other CQAs, the 5’-cap and the 3’-poly(A) tail of mRNA. Although there may be some artifacts as a result of the low abundance of some species, the main capping species was successfully confirmed using this approach within the standard workflow. Future work may offer insights on how to more reliably and comprehensively profile the 5’ cap species. Poly(A) tail characterization is more straightforward because its size allows its chromatographic separation from the other oligonucleotides and analysis via a spectral deconvolution approach.
Routine use of this type of approach in a GMP-compliant environment would benefit from the higher (>90%) sequence coverage yielded by unique digestion products from partial digestion oligonucleotides with overlapping segments of sequence. Automated fragmentation-based sequence confirmation can add value by deciphering sequence isomers for an increment in sequence coverages. Even more so, we found that inclusion of complementary sequence coverages obtained from mapping with a second enzyme is the most effective menas to comprehensively map an RNA molecule.
Acknowledgments
We would like to thank Kaneka Eurogentec and the Region Wallonne for their collaboration and support in the PIT ATMP–Convention 8880 project. We would also like to thank Scott Berger from Waters Technologies Corporation for his critical review of this manuscript.
Waters, RapiZyme, IonHance, Acquity, BEH, Xevo, UPLC, and waters_connect are trademarks of Waters Technologies Corporation. Excel is atrademark of Microsoft Corporation. All other trademarks are the property of their respective owners.
References
(1) Aljabali, A. A. A.; El‐Tanani, M.; Tambuwala, M. M. Principles of CRISPR-Cas9 Technology: Advancements in Genome Editing and Emerging Trends in Drug Delivery. J. Drug Deliv. Sci. Technol. 2024, 92 (105338), 105338–105338. DOI: 10.1016/j.jddst.2024.105338
(2) Gote, V.; Bolla, P. K.; Kommineni, N.; et al. A Comprehensive Review of MRNA Vaccines. Int. J. Mol. Sci. 2023, 24 (3), 2700. DOI: 10.3390/ijms24032700
(3) Weber, J. S.; Carlino, M. S.; Khattak, A. et al. Individualised Neoantigen Therapy MRNA-4157 (V940) plus Pembrolizumab versus Pembrolizumab Monotherapy in Resected Melanoma (KEYNOTE-942): A Randomised, Phase 2b Study. Lancet 2024, 403 (10427). DOI: 10.1016/S0140-6736(23)02268-7
(4) USP, USP Analytical Procedures for mRNA Vaccine Quality (Draft Guidelines)–3rd Edition. https://go.usp.org/mRNAVaccineQuality (accessed 2024-11-13)
(5) Gao, J.; Wu, H.; Shi, X.; et al. Comparison of Next-Generation Sequencing, Quantitative PCR, and Sanger Sequencing for Mutation Profiling of EGFR, KRAS, PIK3CA and BRAF in Clinical Lung Tumors. Clin. Lab. 2016, 62 (04/2016). DOI: 10.7754/Clin.Lab.2015.150837
(6) Roy, S.; Coldren, C.; Karunamurthy, A.; et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. JMD 2018, 20 (1), 4–27. DOI: 10.1016/j.jmoldx.2017.11.003
(7) Vanhinsbergh, C. J.; Criscuolo, A.; Sutton, J. N.; et al. Characterization and Sequence Mapping of Large RNA and MRNA Therapeutics Using Mass Spectrometry. Anal. Chem. 2022, 94 (20), 7339–7349. DOI: 10.1021/acs.analchem.2c00765
(8) Krull, I. S.; Swartz, M. E. Validation and Peptide Mapping. LCGC N. Am. 2007, 25 (5), 468–475.
(9) Goyon, A.; Scott, B.; Kurita, K.; et al. Full Sequencing of CRISPR/Cas9 Single Guide RNA (SgRNA) via Parallel Ribonuclease Digestions and Hydrophilic Interaction Liquid Chromatography–High-Resolution Mass Spectrometry Analysis. Anal. Chem. 2021, 93 (44), 14792–14801. DOI: 10.1021/acs.anal-chem.1c03533
(10) Addepalli, B.; Venus, S.; Thakur, P.; Limbach, P. A. Novel Ribonuclease Activity of Cusativin from Cucumis Sativus for Mapping Nucleoside Modifications in RNA. Anal. Bioanal. Chem. 2017, 409 (24), 5645–5654. DOI: 10.1007/s00216-017-0500-x
(11) Addepalli, B.; Lesner, N. P.; Limbach, P. A. Detection of RNA Nucleoside Modifications with the Uridine-Specific Ribonuclease MC1 from Momordica Charantia. RNA 2015, 21 (10), 1746–1756. DOI: 10.1261/rna.052472.115
(12) Wolf, E. J.; Grünberg, S.; Dai, N.; et al. Human RNase 4 Improves MRNA Sequence Characterization by LC–MS/MS. Nucleic Acids Res. 2022, 50 (18), e106–e106. DOI: 10.1093/nar/gkac632
(13) Menneteau, T.; Butré, C. I.; Mouvet, D.; Delobel A. Multi-Attribute Monitoring of Therapeutic MRNA by Liquid Chromatography–Mass Spectrometry. LCGC Advances in Biopharmaceutical Analysis, Supplement to LCGC Eur. 2023, 36 (s10), 18–24. DOI: 10.56530/lcgc.eu.fd3584v
About the Authors
THOMAS MENNETEAU is an R&D Scientist at Quality Assistance (Donstiennes, Belgium).
BALASUBRAHMANYAM ADDEPALLI is a Scientist Director at Waters Corporation (Milford, USA).
TATIANA JOHNSTON is a Senior Scientist at Waters Corporation (Milford, USA).
CHRISTIAN REIDY is a Product Manager at Waters Corporation (Milford, USA).
MATTHEW GORTON is a Product Manager at Waters Corporation (Wilmslow, UK).
JENNIFER BOUCHENNA is R&D Scientist at Quality Assistance (Donstiennes, Belgium).
NICK PITTMAN is a Marketing Manager at Waters Corporation (Wilmslow, UK).
CLAIRE I. BUTRÉ is an R&D Technical Leader at Quality Assistance (Donstiennes, Belgium).
DAMIEN MOUVET is a Senior Scientific Manager at Quality Assistance (Donstiennes, Belgium).
LAETITIA DENBIGH is a Director Program Lead at Waters Corporation (Wilmslow, UK).
MATTHEW LAUBER is a Senior Director Portfolio Owner at Waters Corporation (Milford, USA).
ARNAUD DELOBEL is an R&D and Innovation Director at Quality Assistance (Donstiennes, Belgium).














. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

