What’s in a Name?
What’s in a Name?
Primary record is a term that was defined by the MHRA (Medicines and Healthcare products Regulatory Agency, the UK drug regulator) in data integrity guidance issued in 2015. In this instalment of Questions of Quality we explore what this term means in practice, and compare it with raw data in the European Union Good Manufacturing Practices (EU GMPs) and complete data in US Food and Drug Administration (FDA) GMPs. Why can’t we have harmonization of terms?
The term “primary record” has been used informally in UK Good Manufacturing Practice for many years. The use of the term refers to the collection of data, metadata, information, and knowledge generated in the course of executing an analytical procedure. The primary record needs to show traceability throughout the procedure from sample management to the reportable result. However, the term is not defined in either the European Union Good Manufacturing Practice (EU GMP) glossary (1) or in the US Food and Drug Administration (FDA) GMP regulations (2). In January and March 2015, the UK regulatory authority, the Medicines and Healthcare products Regulatory Agency (MHRA), published two versions of a guide for industry about data integrity with a list of definitions (3,4). In these documents the MHRA defined the primary record as:
The record that takes primacy in cases where data collected or retained concurrently by more than one method fail to concur.
The FDA, on the other hand, defines requirements for complete data in 21 CFR §211.194(a) (2):
Laboratory records shall include complete data derived from all tests necessary to assure compliance with established specifications and standards, including examinations and assays as follows:…
Surprisingly, there is nothing in EU GMP to define the collection of data and information that constitutes the execution of a single analytical procedure. We believe this constitutes a primary analytical record. The collation of all the primary analytical records for a sample from a manufactured batch would constitute a complete quality control or analytical batch record. Before discussing the issues with regulatory definitions, we would like to delve further into the concept of data, information, and knowledge as applied in a chromatography laboratory.
A Primary Analytical Record Consists of Data, Information, and Knowledge
Any consideration of a primary analytical record should not just focus on data, metadata, and processed records but also the information and knowledge that is extracted or derived from these records. Information and knowledge are used to make decisions and their integrity is just as important as the integrity of the underlying data. There is a hierarchy of data that is transformed to information and information can be aggregated to knowledge as shown in Figure 1. From this we can see that there is considerably more data than information, reflecting the origin of information from the underlying data. There is also more information than knowledge, again reflecting the extraction process. However, one person’s information is another’s data. For example, one can take the cumulative information and knowledge for a specific product batch and use it as data in a product quality review (5).


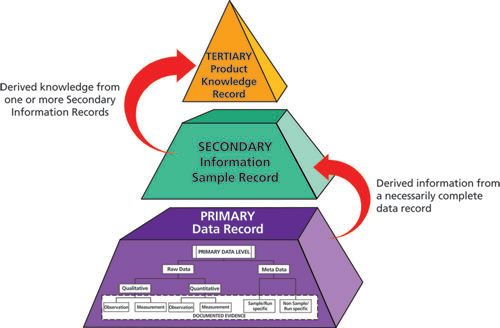
Figure 1: Data, information, and knowledge hierachy.
However, according to the MHRA guidance the definition of data is:
Information derived or obtained from raw data (for example, a reported analytical result)
(4) This definition is misleading. Data cannot equate to information as we can see from Figure 1. Information can be derived or obtained by processing or interpreting data; however, data can never be equated to the information derived. As shown in Figure 3, after the acquisition of data there is a conversion to information (secondary level) and then to knowledge (tertiary level). The figure is also incomplete as we can see from our definition of primary analytical record and that contained in the Principle of EU Chapter 4 (6). Integrity of the whole acquisition, conversion, and retention processes must be assured. Ensuring the integrity of information and knowledge is just as vital as ensuring the integrity of data.
Definition of a Primary Analytical Record
In principle, a primary analytical record must be complete, accurate, and contemporaneous, which will lead to the ALCOA+ criteria that we will discuss later in this column. The GMP traceability criteria of who did what, when, and why is applied throughout the generation and interpretation of the data and to the calculation of the reportable result. These principles should also be applicable to other regulatory disciplines (for example, Good Laboratory Practice and Good Clinical Practice) as well as quality systems such as ISO 9001 and ISO 17025. What do the EU regulations say? In the Principle of EU GMP chapter 4 (6) on documentation the two main document types (instructions and records) are presented. When executed instructions, for example, a standard operating procedure (SOP) or analytical procedure, when executed result in the generation of records or evidence that the procedure has been followed. The chapter also mentions the concept of raw data (6):
Provide evidence of various actions taken to demonstrate compliance with instructions, e.g. activities, events, investigations, and in the case of manufactured batches a history of each batch of product, including its distribution.
Records include the raw data which is used to generate other records. For electronic records regulated users should define which data are to be used as raw data. At least, all data on which quality decisions are based should be defined as raw data.
While Chapter 4 (6) talks about raw data there is no overarching term for an analytical batch record or the collection of data resulting from quality control testing. Chapter 6 on quality control merely requires that test documents pertaining to batch release need to be retained (7). Therefore, in the absence of a suitable definition, we would like to put forward a definition of primary analytical record in two parts as:
A primary analytical record consists of all the data, information, and knowledge input or generated in the course of executing an analytical procedure from the sample to the reportable value to assure traceability and integrity.
The adequacy of an enduring primary analytical record is its demonstrable ability to align with data integrity principles.
This definition works for both hybrid and electronic laboratory computerized systems (heterogeneous and homogeneous systems, respectively, if we use the EU GMP Chapter 4 terminology).
Putting the Primary Analytical Record in Context
Figure 2 illustrates where a primary analytical record sits within a GMP environment. It depicts an overall GMP system; from the regulations through to the interpretation by an individual company to the production of a single batch of product in compliance with the marketing authorization and the company’s quality control system. Two sets of records are produced – the manufacturing record and, for each analytical procedure executed, a primary analytical record. The combination of all analytical procedures executed on the samples from the batch constitutes the quality control records. The combination of the QC record and the manufacturing record constitute the batch record, which is used for release purposes.

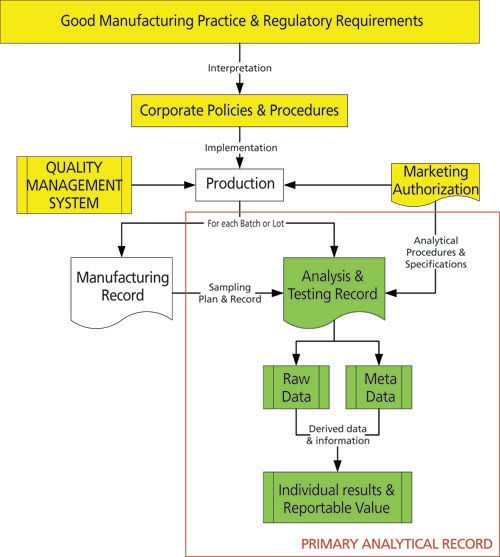
Figure 2: A primary analytical record within a Good Manufacturing Practice environment. Figure 3 shows the elements of a primary analytical record in more detail. The process flow illustrates the execution of an analytical procedure from sample management via analysis through to the calculation of a reportable result that is compared with the product specification. Sample management and sample preparation stages of the analytical procedure are shown in green while the analysis and subsequent processing stages are shown in yellow. The reportable result is in red. Linked with the analysis and processing are the associated metadata such as the audit trail, instrument control, and processing files. Omitted from Figure 3 are the links to the individuals who performed the activities and the time flow, which is implied moving from left to right. The principles presented above can be applied to other GXP regulatory disciplines as well as a quality system such as ISO 17025.

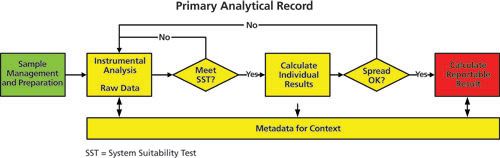
Figure 3: General concepts for a primary analytical record. Figure 3 can be applied to different analytical processes and computerized systems; for example, where the output from an instrument is input into a spreadsheet, or when the data acquired is transferred to an electronic laboratory notebook (ELN) for further calculations. However, regardless of the approach and systems used any primary record must comply with data integrity principles. Data integrity principles are listed under the acronym ALCOA+ and are presented and discussed below: •
Attributable:
Identification of the individual who performed an activity. •
Legible:
Can you read the electronic data together with any associated metadata, or all written entries on paper? Legible should also extend to any original data that has been changed or modified by an authorized individual so that the original entry is not obscured. •
Contemporaneous:
Documented (on paper or electronically) at the time of an activity. •
Original:
A written observation, printout or a certified copy thereof, or an electronic record including all metadata of an activity. •
Accurate:
No errors in the original observation(s) and no editing without documented amendments/audit trail entries by authorized personnel. •
Complete:
All data from an analysis including any data generated before a problem is observed, data generated after part of the work or all of the work is repeated or reanalysis is performed on the sample. For hybrid systems, the paper output must be linked to the underlying electronic records used to produce it. •
Consistent:
All elements of the primary analytical record such as the sequence of events are in order and do not contradict each other. Data files are date (all processes) and time (when using a hybrid or electronic systems) stamped in the expected order. •
Enduring:
Recorded on authorized media (laboratory notebooks, numbered worksheets) for which there is accountability, or electronic media that can last throughout the record retention period. •
Available:
The complete collection of records can be accessed or retrieved for review and audit or inspection over the lifetime of the record. The FDA (ALCOA) initially developed these first five criteria; the additional criteria were added by the European Medicines Agency when discussing integrity criteria for electronic source data from clinical studies (ALCOA+) (8).
Primary Analytical Record and Complete Data
Our definition of primary analytical record specifies that it must contain all the data generated in the course of executing an analytical procedure, assuring traceability from sample to reportable value. The primary record comprises the original records, not copies, in the system where they are created. The alignment of primary analytical record with complete data means that you can comply with EU and FDA GMP regulations with a harmonized approach. Complete data has been discussed in two previous Questions of Quality instalments (9,10). A flow chart was presented showing the types of records that were created when undertaking a chromatographic analysis. We will update this to illustrate what a primary analytical record means in practice.
Primary Analytical Record in Practice
Figure 4 shows the analytical process flow carried out by a chromatography data system (CDS) by way of an example of a primary analytical record. For the purposes of discussion, all of the work outlined in this Figure takes place within a CDS. There is no interface with another application or spreadsheet to undertake any part of the workflow other than the sample management, including preparation for analysis.

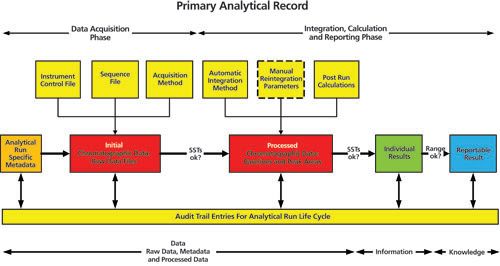
Figure 4: Interpretation of a primary analytical record for chromatographic analysis using a CDS. It is worth noting that the example focusses on chromatography data but the principles apply to any other analytical process or computerised system. Figure 4 is divided into two phases: the first phase is data acquisition, which is followed by the second phase of integration, calculation, and reporting. In addition, at the bottom of the figure are three sections dealing with data, information, and knowledge that link back to Figure 2. This illustrates our concept of the conversion of data to information and then to knowledge. System suitability tests (SSTs) refer to system suitability test samples that are used as a point of use check that the chromatographic system is under control before and during the analytical run. The SSTs are there to ensure that the data generated is within the designated analytical control strategy of the method.
Data; The Foundation of Information and Knowledge
Data Acquisition Phase:
What are the analytical pre-requisites required and what data is generated during the data acquisition phase of the primary analytical record? Data acquisition within a CDS needs the following files or metadata: • A data acquisition file which specifies the parameters to collect the data for each injection for example, sampling rate, run time, etc; • A sequence file to define the order of injections with injection volume. Run-specific metadata can be entered here such as sample identities, number of aliquots, dilutions, purities of standards etc; • An instrument control file for operating parameters such as isocratic or gradient profile, flow rate, oven or autosampler injection temperatures, detector wavelength(s), or scan rate; • Run-specific metadata such as sample identity, batch or lot number, sample preparation data for example, weights, reference standards and their purities, dilutions. This information may be generated outside of the CDS but can either be entered manually by a user or transferred electronically from another computerized system. • An audit trail to identify the individuals involved in each phase of the work together with date and time stamp of the activity. These files do not function in isolation and they enable the CDS to acquire the initial chromatographic data files, which should be defined as raw data as per the Principle of EU GMP Chapter 4 (6). Entries and their modifications are documented in the audit trail for the run, which are part of the contextual metadata for the analytical procedure.
Integration, Calculation, and Reporting Phase:
Before starting this phase there can be a visual or automated check to confirm that the SSTs are acceptable (peak shape and apparent area) (Figure 4). In this phase, the raw data are processed by the CDS software to provide the required information in a pre-defined manner: • Integration parameters are applied to all the acquired chromatography data files of the run. These parameters are applied automatically (11,12) to integrate the peaks, measure the individual peak areas, and identify the analytes of interest. • Manual integration is an option that could be applied if laboratory procedures permit. This was discussed in the last Questions of Quality instalment, which differentiated between manual intervention of the overall integration parameters and manual integration where a chromatographer manually positions the baselines of peaks (12). • Post-run calculations (SST calculations and assessment versus acceptance criteria, peak area calculation followed by calculation of the amounts of analyte in each aliquot after adjustment for any weight, purity, dilution, or other factors) can be applied after the integration. • An audit trail to identify the individuals involved in each phase of the work together with date and time stamp of the activity. The end result of this is processed raw data and, as noted in EU GMP chapter 4 (6), raw data generates other records. We now have peak areas and SST results. In addition, all manual interventions by an operator will be recorded in the CDS audit trail for the run – more metadata within the umbrella of the primary analytical record.
Information
We now enter the information domain, which is presented in Figure 1 as the calculation of the amount or concentration of analyte(s) in the injections of an individual preparation. For example, if a sample has two weighed aliquots taken through an analytical procedure and each preparation is injected twice (or two vials are prepared with a single injection of each) there will be four individual results for that sample. This represents the information in the sample and is derived from the underlying data shown in Figure 2. This information also forms part of the primary analytical record.
Knowledge
The individual results are combined in a pre-determined manner, usually by averaging, to produce a final reportable result that is compared with the specification. This is the knowledge element of the primary analytical record.
Traceability
Throughout the creation of a primary analytical record there must be explicit linkage to key items to support data integrity such as identification of: • Individual analytical personnel and their actions that are accurately time and date stamped; • Samples analysed, which are then linked to the material batch numbers or a stability or study plan; • Instruments used to generate data (ideally noting that they are qualified); • Name and version numbers of software used to acquire and process data and report results; • Linkage to the raw data files and metadata from the analysis; • The analytical procedure and version used to acquire data.
Common Elements
Throughout the whole process there are some common elements that need to be highlighted for a CDS as well as other systems: • A primary analytical record is not a single record but a collection of data, metadata, information, and knowledge for comparison with a specification. • Audit trail entries are acquired throughout the initial data acquisition and the conversion of data to information and then to knowledge (either within a single system as shown in Figure 4 or across systems if more than one are involved). • Figure 4 illustrates the generation of raw data and the process of creating other records as data is processed and converted to knowledge. This illustrates the principle of EU GMP Chapter 4 (6) that: Records include the raw data, which is used to generate other records.
MHRA and the Alternative Definitions Contrasted
The two MHRA guidance documents (3,4) contain a definition of primary record together with an expectation that is shown in Table 1. It is good to see that the MHRA is proactive and has made modifications to their document in light of feedback received.

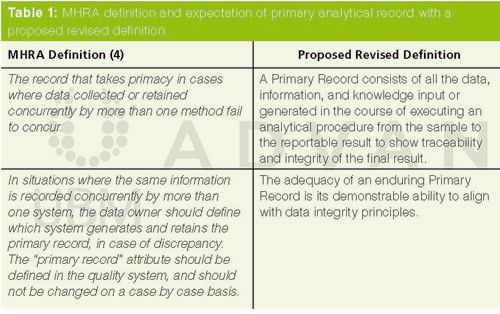
The MHRA document raises a question regarding the practicality of their definition of primary record, namely: concurrent. It is difficult to see a situation in any regulated laboratory where two tests for the same parameter would be conducted. The
proposed revised
definition fills a gap in EU GMP by defining a collection of analytical data, information, and knowledge associated with the analysis of a sample by a specific analytical procedure. It takes the raw data and combines all records and metadata created or updated in the subsequent processing to the individual values and finally the integrity of the reportable result.
Summary
This column looked at the MHRA definition of Primary Record and proposed an alternative. The alternative definition is closer in meaning to complete data in US GMP regulations and is a potential means of using a harmonized approach for complying with EU and FDA GMP.
Acknowledgements
The authors would like to thank Monica Cahilly, Mark Newton, Lorrie Schuessler, and Paul Smith for their comments and advice in preparation of this article.
References
1. European Commission Health and Consumers Directorate-General,
Eudralex: The Rules Governing
Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products
for Human and Veterinary Use
; Glossary (Brussels, Belgium, 2001). 2. US Food and Drug Administration,
21 CFR 211, Current Good Manufacturing Practice regulations for Finished Pharmaceutical Products
(FDA, Rockville, Maryland, USA, 2008). 3. MHRA GMP Data Integrity Definitions and Guidance for Industry (January 2015). 4. MHRA GMP Data Integrity Definitions and Guidance for Industry (March 2015). 5. European Commission Health and Consumers Directorate-General,
Eudralex: The Rules Governing
Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products
for Human and Veterinary Use
; Chapter 1 Pharmaceutical Quality Systems (Brussels, Belgium, 2013). 6. European Commission Health and Consumers Directorate-General,
Eudralex: The Rules Governing
Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products
for Human and Veterinary Use
; Chapter 4 Documentation (Brussels, Belgium, 2013). 7. European Commission Health and Consumers Directorate-General,
Eudralex: The Rules Governing
Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Medicinal Products
for Human and Veterinary Use
; Chapter 6 Quality Control (Brussels, Belgium, 2014). 8. EMA Electronic Source Data, 2007. 9. R.D. McDowall,
LCGC Europe
26
(6), 338–343 (2013) 10. R.D. McDowall,
LCGC Europe
28
(7), 389–392 (2013). 11. N. Dyson,
Chromatographic Integration Methods
(Second Edition, Royal Society of Chemistry, 1998). 12. R.D. McDowall,
LCGC Europe
28
(6), 336–342 (2015).








New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.

Quantifying Terpenes in Hydrodistilled Cannabis sativa Essential Oil with GC-MS
April 21st 2025A recent study conducted at the University of Georgia, (Athens, Georgia) presented a validated method for quantifying 18 terpenes in Cannabis sativa essential oil, extracted via hydrodistillation. The method, utilizing gas chromatography–mass spectrometry (GC–MS) with selected ion monitoring (SIM), includes using internal standards (n-tridecane and octadecane) for accurate analysis, with key validation parameters—such as specificity, accuracy, precision, and detection limits—thoroughly assessed. LCGC International spoke to Noelle Joy of the University of Georgia, corresponding author of this paper discussing the method, about its creation and benefits it offers the analytical community.


