Top 10 HPLC Column Myths
LCGC North America


In any field there are often "misconceptions" or "myths" that are perpetuated and passed on to the next generation.
Myth 1: You Can't Reverse an HPLC Column
False. In practice, a high performance liquid chromatography (HPLC) column is packed at pressures much higher (often as much as twofold) than its maximum operating pressure. Thus, if the proper slurry solvent was used and the time allotted for the packed bed to be stabilized, a well-packed column should be able to work in both directions. Some reasons why one would want to flow a column in the opposite direction include backflushing operations in column switching, rinsing a column with stubborn strongly sorbed sample at its entrance (a shorter pathlength than to flush the insoluble materials through the entire column length), and flushing trapped particulates to decrease pressure buildup.



Ronald E. Majors
There is one exception in reversing the flow of an HPLC column. If the manufacturer has used a higher porosity frit at the entrance to the column, by reversing the column it might be possible to flush particles out of the packed bed. When the column is packed at the factory, the porosity of frit at the outlet of the column must be lower than the smallest particle size of the column. For example, if the column packing has an average diameter of 5 μm and a particle size distribution of 3–7 μm, the frit used at the column exit must be less than 3 μm so that there is no possibility that particles will escape the packed bed when the column is being used. For that case, most manufacturers choose a 2-μm frit.
Why would a manufacturer have different porosity frits at the column inlets and outlets? Simple. A higher porosity frit has fewer tendencies to plug than a lower porosity frit. A 0.5-μm frit will plug faster than a 2-μm frit. So, to prevent rapid pressure buildup and customer complaints, a manufacturer might use a more forgiving, larger porosity frit at the inlet. Usually, the column will be marked with an arrow that indicates that it should be used in one direction only. Before reversing an HPLC column, it is best to consult the column instruction sheet or check with the column manufacturer to see if the column can be reversed.
Myth 2: All C18 (L1) Columns Are the Same
False. In the early days of HPLC, octadecylsilane (most often referred to as an ODS or C18 phase) was one of the first bonded stationary phases that became available for the new technique called "reversed-phase chromatography." It became the standard phase for reversed-phase chromatography and quickly was adopted by most practitioners. Because the pharmaceutical industry was an early adopter of HPLC and regulatory bodies did not want to bless a particular manufacturer's brand of column, the FDA and USP developed a classification system that gave a generic designation for each new method that was submitted under a new drug application. For HPLC columns, an "L" designation was given and because C18 was used in a majority of the submittals, it became "L1." As additional phases were added, they were given their own "L" number (for example, L7 C8, L10 CN, L11 Phenyl, and so forth).
Unfortunately, this designation system proved to be unreliable because each commercial C18 column was synthesized differently using silica gel as the base material and the resulting reversed-phase columns had different properties. For example, some manufacturers used octa-decylmonochlorosilane and a low surface area silica gel (Figure 1) while others used the same silane but bonded it to a higher surface area silica gel. These two C18 columns would behave differently with the latter having more C18 phase than the former. A lower coverage bonded phase sometimes had unreacted silanols that caused mixed retention mechanisms. Some manufacturers use di- and trichlorosilanes and polymerized the bonded phase to form a thicker layer with different diffusional properties. To cut down on unreacted silanols that remained after the bonding process, some manufacturers endcap these silanols with a small silane (for example, trimethylmonochlorosilane). Silanols are sometimes responsible for tailing of basic compounds at intermediate pH values. Some manufacturers even went further and double-endcapped with a second small silane to provide a more inert surface. Some manufacturers used polymeric base materials and then bonded C18 moieties to their surface resulting in a totally different C18 packing material, yet it was still considered an "L1." Others use organoalkoxysilanes that display a different degree of reactivity and can produce a slightly different C18 bonded phase than would an organochlorosilane reagent.

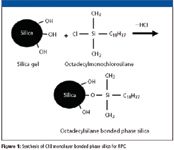
Figure 1
So, the statement "a C18 is not a C18 is not a C18" holds true even today. All C18 phases are not alike and the user should make sure that once a method is developed, he or she sticks with the same column part number to ensure rugged and robust methods. In recent times, Dolan and Snyder (1,2) have studied a large number of C18 phases in an attempt to determine which phases have similar retention and selectivity features. By characterizing the phases based upon probes with as many as five retention parameters, they have classified phases with a numerical value so that one might be able to choose the closest phase in performance characteristics in case the original phase is not available from a particular manufacturer. Their approach also allows one to find a C18 phase totally unlike their existing phase that can aid in column selection during method development.
Myth 3: Guard Columns Do Not Affect a Separation
False. First of all, having a guard column is a good idea. However, the choice of a guard column can have a great effect on a separation if the wrong configuration or phase is chosen. Remember, the purpose of a guard column is to protect the analytical column against fouling by highly retained sample components, particulates, and other undesirable materials. The guard column is much less expensive than the analytical column that it protects. Thus, it can be replaced more often rather than the more expensive analytical column.
Ideally, the guard column chosen should have the exact same stationary phase as used in the analytical column. If the phase is more retentive (for example, has a greater carbon loading or is a mixed-mode phase), then it can affect the separation in a detrimental manner by causing retention time shifts or even selectivity differences. If the phase is less retentive, it might or might not cause a problem unless the phase chosen impacts the overall selectivity.
For the guard column to have minimal impact on separation performance, it must be properly plumbed into the flow stream. Obviously, the guard column is inserted between the injector and the analytical column but if too much connecting tubing (either too long or two large of an internal diameter) is used, extracolumn band spreading can occur that can affect the overall separation. Systems employing integrated guard columns where the guard column literally butts up against the analytical column would offer the best solution. However, integrated guard columns usually are associated with cartridge column systems, which seem to have come into disfavor. Whatever the configuration, the guard column should be able to be removed easily and replaced from the HPLC system without affecting its operation.
Theoretically, if the guard column is adding additional column length to the analytical column, there should be additional plates generated that should increase the chromatographic resolution. However, due to some of the factors discussed previously, often the addition of a guard column just maintains the separation at best and sometimes detracts from it at worst. The advantage of the guard column is increasing column life and not necessarily increasing the overall efficiency.
Myth 4: High Temperature Always Leads to Better Separations
False. As the temperature increases, the viscosity of the mobile phase decreases and therefore the rate of solute mass transfer should increase thereby offering better chromatographic efficiency. True, but besides the column efficiency term (H), temperature also can affect the retention factor (k) and selectivity (α). The impact on these terms can result in improved resolution (which is really what we are concerned about in chromatography) or detract from it. Retention usually decreases as the temperature increases because, being a thermodynamic parameter, the analyte prefers to remain in the mobile phase and are eluted sooner from the column. However, different chemical species might have different degrees of change in their retention versus temperature. Stated more correctly, their van't Hoff plots (ln k vs. 1/T where the temperature T is measured in kelvins) might display different slopes; in other words, their α values can change. In addition, high temperature can cause a low k peak to be eluted so quickly that it can be eluted at or near t0, the unretained peak retention time, and therefore be difficult to quantitate.
Take the example of analgesics shown in Figure 2. This series of chromatograms shows the separation of seven analgesics at column temperatures of 20–90 °C. One can note several features of the chromatograms. First, the retention of all peaks is decreased with higher temperatures and the peaks do become narrower indicating improved efficiency. Second, one of the analgesic compounds, salicylic acid, has a much greater retention change with temperature relative to peaks 5 and 6, the closest eluted compounds. In fact, the elution order is reversed after increasing the temperature from 20 to 40 °C. At the intermediate 30 °C temperature, the salicylic acid and peak number 6, phenacetin, are coeluted. So in this case, temperatures greater than 40 °C resulted in a shorter separation time for the entire separation yet the elution order was changed from the lower temperatures.


Figure 2
Of course, a side benefit of operating at higher temperature is the decrease in column operating pressure that allows one to use higher flow rates or smaller particles.
Another experimental parameter that can cause column performance problems at higher temperature is the possible thermal mismatch of the incoming mobile phase. For example, if the column is heated to 60 °C and the incoming solvent is at room temperature, the incoming cooler solvent can cause peaks to be distorted because of the differences in the temperature that the solutes can experience in the initial part of the column. It is recommended that one preheats the mobile phase when using higher column temperatures.
Myth 5: The Higher the Carbon Load the Better the Reversed-Phase Column
False. There appears to be misconceptions about the role of chain length, carbon loading, surface coverage, and so forth when it comes to the popular alkyl bonded phases. In general, for a truly reversed-phase mechanism where retention is based upon the relative hydrophobicities of analyte molecules, retention is usually based upon the carbon load. The higher the carbon load, the more the retention. Carbon load can be proportional to the chain length but not necessarily so. A typical silica gel has about 8.0 μmol/m2 of reactive silanols that are used for bonding organosilane reagents. If, for example, for a given surface coverage of a monolayer bonded phase in micromoles per meter squared, the longer the chain length, the more carbon would be present and therefore retention would be proportional to chain length. However, if a shorter chain bonded phase (say, a C8) had a higher surface coverage resulting in more carbon on the surface, then it could have more retention than a C18 bonded phase. In addition, some manufacturers use di- and trichloro-organosilanes where polymerization is used to increase the phase coverage; a shorter chain length polymeric phase could conceivably have a much greater coverage than a longer chain monomeric phase. Sometimes these polymeric phases have a thicker layer of the bonded phase that results in poorer stationary phase mass transfer than for a monomeric phase.
Very highly loaded reversed-phase columns also are prone to phase collapse or more correctly stationary phase dewetting (3). For such columns, when the amount of organic modifier falls below 10% in an aqueous environment, these "oil-like" hydrophobic phases tend to prefer to self-associate rather than being in a polar aqueous solution (that is, like prefers like). So, in this situation, having a high carbon load might be detrimental to successful reversed-phase chromatography and reproducible retention time.
Myth 6: One Always Gets More Efficiency Using a Column Packed with Smaller Particles
False. Although, for a well-packed column, the efficiency increases as the average diameter of the particles in a packed column decreases, if the extracolumn contributions to band broadening of a particular HPLC hardware system are sufficiently high then the full performance of this column will not be realized. Extracolumn contributions consist of all the additional volume outside of the packing itself. These contributions include the following volumes:
- Injection volume including the loop size as well as the additional volume within the injection valve.
- Tubing volume from the injector outlet to the column inlet.
- Guard column interstitial volume including endfittings (if present).
- In-line filter volume (if present).
- The internal volume within the inlet column fitting including porous frit and flow channels.
- The interstitial column volume.
- The internal volume within the bottom column fitting including porous frit and flow channels.
- Tubing volume from the column outlet to the detector inlet.
- Tubing volume within the detector before the flow cell.
- Flow-cell volume.
The variances of all of these volumes are additive. Thus, one can minimize injection volume and keep the column inlet tubing of short length and small internal diameter but if there is a 1-m piece of 0.20-in. connecting tubing from the column outlet to the detector, then the band broadening might be large enough to effect the overall efficiency of the separation. So, make sure that the extracolumn effects are minimized if you expect to get the expected efficiency of a sub-2-μm HPLC column or a 1.0-mm i.d. microbore column. For more information on proper column and hardware selection, see reference 4.
Myth 7: For Silica Gel Packings, Residual Silanols Are Responsible for Tailing
False. Under certain circumstances, silanols, present on all silica-based bonded phase columns, can cause tailing, especially with basic compounds. This tailing is ascribed to the fact that the silanol group is a weak acid with a pKa of approximately 4.5–4.7. Thus, as the pH of the mobile phase approaches the 4–5 range the silanol becomes ionized and can interact with positively charged molecules, such as a protonated amine, by electrostatic attractions. This interaction can be minimized by suppressing the ionization of the silanol by lowering the pH to less than 3. For some basic compounds, tailing can sometimes occur at lower pH values. There are specialized reversed-phase packings that are especially designed to provide good peak shape for basic compounds.
There are three basic causes of peak tailing: chemistry problems (one of which was discussed previously); column packing problems; and instrument hardware problems. All three can cause peak tailing and one must investigate the root causes to determine a remedy. It is outside the scope of this question to explore all three in detail but a few examples of each type will give you the general idea. First, in addition to interaction with silanols, chemistry-related tailing problems can manifest themselves in several ways. Metal chelating compounds can interact with trace metals in the column packing and was particularly noticeable for older silica materials. Employing the wrong injection solvent can sometimes lead to tailing. Injecting the sample is a much stronger solvent than the mobile phase can give peak distortion. Tailing can result from built-up material from strongly retained sample components or mobile phase impurities at the head of the column. This material can act as a different stationary phase, causing tailing problems for compounds that interact. On occasion, mixed modes can occur thereby causing tailing. Here solutes can interact by reversed-phase and ionic interaction with active groups. Sometimes, when the mobile phase pH is wrong and samples are partially ionized, the resulting peaks can be distorted and resemble tailing. A small partially coeluted peak on the backside of a larger peak can sometimes look like tailing.
Column packing problems can cause peak tailing. A void at the head of a column can lead to doublet peaks or peak tailing. Columns that are not well packed can have channeling resulting in bad peak shape. If the column is overloaded by too great of a sample mass, tailing can occur, although peak fronting is more prevalent. At low injection masses, tailing can occur by overloading silanols.
Let us consider a few instrumentation and hardware problems that might show up as tailing. Tailing can be caused by extracolumn effects and other unswept volumes in the flow lines. Unswept volumes from an endfitting or an improper connection can manifest itself as peak tailing. Pressure pulses from the injector can cause columns voids that can give rise to tailing. A slow detector time constant with quickly eluted peaks can give peak broadening sometimes resembling tailing. Thermal mismatch between the incoming solvent at room temperature and a high column operating temperature also can cause peak distortion.
So, it is not always the silanols that cause peak tailing in HPLC.
Myth 8: Silica Gel–Based Packings Can Be Used Only From pH 2 to 7
False. Generally, there are two mechanisms of chemical deterioration of an HPLC reversed-phase column: catalyzed hydrolysis of siloxane bonds at pH less than 2; and dissolution of the silica gel by hydroxide ion at pH values greater than 7–8. These two phenomena have been extensively studied by Kirkland and coworkers (5–7). At pH values less than 2, the -Si-O-Si- (siloxane) bond can be attacked by hydronium ion (H3O+ ) cleaving the bonded phase, which results in a loss of organic. With time, retention will usually decrease as the carbon content is reduced. It is especially noticeable with the short chain endcapping moieties such as trimethylsilane. The longer chain C18 phases offer some protection of the underlying siloxane by virtue of their steric hindrance but eventually even these will be attacked, especially if the temperature is higher than ambient. There are sterically protected and densely coated bonded phases that help to impede the cleavage of bonded phases on silica gel. Polymeric phases do not show this instability but have the disadvantage of lower efficiency than silica-based phases.
On the high pH side, there must be some way to block the underlying silica gel from attack by hydroxide ion. Once the dissolution process occurs, the column will eventually fail, often developing a void. Specialty columns are made for high pH operation including bidentate C18, hybrid, and polymer-coated silica. These phases chemically protect the base material from hydroxide attack. These packings are capable of withstanding pH values in the 11–12 range. High temperatures should be avoided with most silica-based packings when operating in this range. Of course, polymeric phases can be operated at pH values as high as 13, sometimes even 14, but, as pointed out previously, they have the disadvantage of lower efficiency than silica-based phases.
So, the real answer: special silica-gel bonded phases can operate outside of the pH 2-7 window but care should be exercised with regular silica gel-based packings, especially at high temperature.
Myth 9: Modern HPLC Columns Should Withstand at Least 1000 Injections
False. There are a number of factors that control the number of injections any modern HPLC column can withstand. Some of the factors are based upon the mode: reversed phase, ion exchange, size exclusion, normal phase, chiral, hydrophilic interation, and so forth. Some of these factors are based upon the type of packing in the column — silica gel-, hybrid-, zirconia-, or polymeric-based packings, or a soft gel or degree of crosslinking of resin. Some factors are based upon the phase itself: coverage, type of bond, polymeric, monolayer, or coated phase. Other factors are based upon the operating conditions: pH, temperature, mobile phase constituents, buffer composition, flow rate, pressure, and so on. Still other factors are based upon the sample that is injected: standards only, sample cleanliness, sample pH, sample volume, sample impurities present, solute molecules themselves, and so on.
If a column is abused such as by using it outside of its recommended pH limits or flow-rate range, it might only yield 50 injections or even less. If the sample is simple with no highly retained impurities, then the column might withstand 5000 injections or more. If the column is not continuously operated at its upper limit, it will live longer. If the column is subjected to a variety of samples and never flushed to remove strongly retained impurities, then its life will be shortened.
In the author's experience of visiting many pharmaceutical companies, most of them expect at least 1000 injections out of a 5-μm reversed-phase column when it is used for analyzing formulations, simple drug mixtures and standards. If the column is used for "dirty samples" such as biological fluid extracts or environmental extracts that have not been exhaustively cleaned up, then one should not expect 1000 injections.
So, the number of injections expected is not cut and dry but is highly dependent upon the type of column, operating conditions, sample cleanliness, and degree of abuse. Of course, with guard columns and, perhaps, in-line filters, column life should be longer. In informal polling of audiences during numerous seminars by the author, a small percentage (less than 15%) of the users actually track the number of injections for a given column. So, may chromatographers don't really know how many injections that they get per column lifetime. Some modern HPLC instruments have built-in column modules and software that permits one to monitor the injection number.
Myth 10: Columns Always Should Be Capped Tightly to Prevent Packing Damage from Contact with the Atmosphere
False. In general, the tiny hole in the endfitting, which is probably not bigger than 0.02 in. in diameter, has such a small cross-sectional area that damage to the column resulting from air coming in contact with the packing and the solvent inside the column evaporating is minimal. Even if a small amount of air entered the column, it would probably have a hard time diffusing through the microparticulate packed bed and coming in contact with enough packing material to cause detrimental effects. If there was a small amount of air in the column, as soon as it was pressurized when it was next installed into the HPLC instrument and mobile phase pumped through it, the small amount of air would probably dissolve at high pressure or be flushed out in the initial fluid in a short time and not cause any problems with later use. However, if you would feel more secure by capping the endfittings by all means do so. Most columns come equipped with male compression fitting caps that can be tightened to prevent any possibility of storage solvent evaporation or air entering the packed bed.
Ronald E. Majors "Column Watch" Editor Ronald E. Majors is a Senior Chemist, Columns and Supplies Division, Life Sciences and Chemical Analysis Group, Agilent Technologies, Wilmington, DE, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Sample Prep Perspectives," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com
References
(1) L.R. Snyder and J.W. Dolan, LCGC 22(12), 1146–1152 (2004).
(2) L.R. Snyder and J.W. Dolan, LCGC 23(2), 118–127 (2005).
(3) M. Przybyciel and R.E. Majors, LCGC 20(6), 516–523 (2002).
(5) J.J. Kirkland and J.W. Henderson, J. Chromatogr. Sci. 32, 473–480 (1994).
(6) H.A. Classens, M.A. van Straten, and J.J. Kirkland, J. Chromatogr. 728, 259 (1996).
(7) J.J. Kirkland, LCGC 14(6), 486–500 (1996).















Characterizing Polyamides Using Reversed-Phase Liquid Chromatography
May 5th 2025Polyamides can be difficult to characterize, despite their use in various aspects of everyday life. Vrije Universiteit Amsterdam researchers hoped to address this using a reversed-phase liquid chromatography (RPLC)-based approach.



New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

