Tips, Tricks, and Troubleshooting for Separations of Biomolecules, Part 1: Contemporary Reversed-Phase Protein Separations
LCGC Asia Pacific
Several new materials and columns have been introduced in recent years for reversed-phase separations of proteins. How do I know which one to choose, and which separation conditions will be best for my protein separation?
Several new materials and columns have been introduced in recent years for reversed-phase separations of proteins. How do I know which one to choose, and which separation conditions will be best for my protein separation?
Over the past decade, there has been tremendous growth in the development of biologics for the purpose of treating diseases ranging from cancer to inflammatory disorders such as ankylosing spondylitis. Currently, the majority of these biologics are proteins, and many of them are monoclonal antibodies. This increase in development activity has, in turn, resulted in a dramatic increase in the number of researchers engaged in protein separations-perhaps more than any other time in history. For those who learned the principles of chromatography by doing experiments involving small molecules, the increased importance of protein separations in the chromatography community presents both tremendous opportunities and challenges. Whereas particle and column technologies designed for smallâmolecule applications have steadily improved over the past 20 years (for example, with the refinement of superficially porous particles and development of high-performing stationary-phase chemistries), until recently there has not been as much development of materials for large-biomolecule separations, and many users are still using column technologies developed more than 20 years ago. This lag in development is both an opportunity-advances in materials technologies will inevitably improve separation performance for large biomolecules-and a challenge, because many of the best practices relevant to these separations will have to be rewritten as particle and column technologies evolve.
In my laboratory, we made a major shift about four years ago from focusing mainly on smallâmolecule separations (for example, environmental contaminants and forensic applications) to focusing more than 75% of our effort today on large-biomolecule separations. As I find myself saying often these days, shifting from smallâmolecule to large-molecule separations is not as simple as just injecting different samples. Researchers with a lot of experience with large biomolecules are painfully aware of this difficulty, but chromatographers who are transitioning from smallâ to largeâmolecule analysis may not appreciate the importance of seemingly minor details for achieving high-quality biomolecule separations.
For this instalment of “LC Troubleshooting”, I have asked two of my collaborators in the biomolecule application space to join me in writing about some of the details that we have found to be particularly important to reversed-phase separations of proteins.
Dwight Stoll
Why Do Proteins Behave So Differently?
Perhaps the simplest view of why proteins behave so differently from small molecules in chromatographic systems has to do with their (sometimes very) large size. Whereas a small molecule like ibuprofen has a mass of about 300 Da, proteins are on the order of 20- to 1000-fold larger; monoclonal antibodies (mAbs), which currently dominate the list of top-selling biologic therapeutics, have a mass of roughly 150,000 Da. The large size of these proteins leads to much slower diffusion in solution. They are composed of thousands of atoms, tens of different functional groups, and are sometimes quite reactive. They are invariably produced by living organisms (for example, bacteria or mammalian cells), unlike small molecules, which are generally synthesized from simple starting materials. Subsequent purification steps, which can be very good, are not perfect, meaning that the analyte we are interested in may be chemically heterogeneous. Finally, these large molecules can adopt secondary, tertiary, and even quaternary structures (that is, shapes) that may give rise to interactions with chromatographic media that are difficult to understand.
In the following sections, we have summarized what we and others have learned about how to work effectively with these proteins under reversedâphase conditions. Some of the issues we discuss are better understood at a fundamental level than others, and many laboratories around the world are actively engaged in trying to understand them better. In most cases, there is both a lot we can learn from older literature (1), and a lot that remains to be discovered as we experiment with new technologies being introduced by manufacturers (2).
The Pore Size of the Chromatographic Media Is Very Important
One of the fundamental concepts we learn about liquid chromatography (LC) early on is that diffusion of analytes into and out of the pores of porous particles is important, and when it is too slow, it leads to broadening of chromatographic peaks. Whereas the pores of typical porous particles (nominally 10 nm [100 Å] in diameter) are large enough to accommodate a 300 Da molecule without significantly hindering its diffusion into and out of the pores, these pores are simply not big enough to accommodate a protein. In the best case, the diffusion of the protein will be obstructed, leading to peak broadening. In the worst case, the proteins can effectively become “stuck” inside the particle, leading to serious peak tailing and symptoms that could be interpreted as analyte carryover from one analysis to the next.
Column manufacturers are aware of this problem, of course, but from the perspective of the analyst one of the most important practical questions is “How big is big enough?” In the limit of very large pores, the mechanical stability of the porous particle will be compromised, and the surface area of the stationary phase will be decreased to the point where mass overload (that is, increased peak width that results from injecting too much analyte mass [3,4]) becomes a serious problem. In this way, solving one problem can create a different one if not done carefully.
Figure 1 shows a comparison of the peak shapes observed for the two model proteins bovine serum albumin (BSA, 66 kDa) and myoglobin (17 kDa) obtained under reversedâphase conditions using columns prepared with particles having two different nominal pore diameters-one 160 Å, and the other 300 Å.


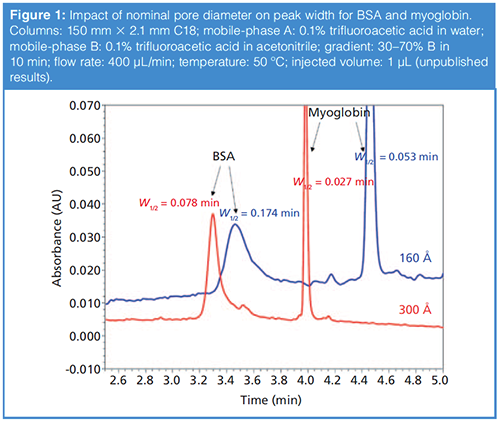
The pore diameter clearly has a profound impact on the peak width. For myoglobin, the peak width decreases by about 50% when the pore diameter is increased from 160 Å to 300 Å. For BSA, which is more than twice the size of myoglobin, the improvement is even better, with a decrease in peak width of about 56%. We note here that the retention also drops significantly with the wider pore diameter material, presumably because of the lower surface area of the particle. This kind of improvement in peak width, which is even more dramatic for larger proteins such as mAbs, has motivated manufacturers to introduce wider pore materials in the 400–500 Å range, and even up to 1500 Å. The most reliable way to determine which particle will perform best for a particular protein is to experimentally measure peak width and retention time. When making these measurements, it is important to make them over a range of injected masses (for example, from 0.1 to 10 µg protein injected) because these data can be used to assess the compromise between pore size and stationaryâphase surface area. Of course, it is not always practical to make these measurements; readers interested in more detail on this topic, or looking for guidance without having to make their own measurements, are referred to a number of recent articles on this topic (3–7).
Column Temperature Is Important, Too
The temperature of the column and mobile phase used during reversedâphase separations of proteins is important as well. There is a compelling case for increasing the temperature well above ambient, because this increased temperature increases the molecular diffusivity of proteins in solution, which can mitigate peak broadening that occurs as a result of slow mass transfer. This approach can improve the efficiency and peak capacity of protein separations in general, and can also be used to improve separation speed through the reduction in mobile-phase viscosity at higher temperatures (4,8). On the other hand, using a temperature that is too high-especially with the acidic mobile phases commonly used for reversedâphase separations of proteins-can cause hydrolysis of the siloxane bond that tethers stationaryâphase ligands to the silica surface in the case of silica-based phases. Advances in stationaryâphase chemistry over the years have considerably improved the chemical stability of some silica-based phases at low pH (4,9). Finally, prolonged exposure of protein analytes to these conditions can also cause on-column degradation of the protein. So, as was the case with pore size discussed above, the question here, too, is “How hot is hot enough?”
First and foremost, users should abide by the guidance of the column manufacturer with respect to the prescribed temperature limits for the column. Then, we consider the following questions:
- Recovery of protein from the column-in other words, does everything that we inject come out within the analysis time?
- Stability of the protein during the separation-is there any detectable degradation inside the column?
Although we don’t believe the cause of incomplete recovery is well understood, what we do know from experiments is that using column temperatures that are too low can lead to incomplete recovery of the protein from the column, and that increasing the column temperature improves the recovery. Figure 2 shows a compelling example of this effect from our own work in the case of reversed-phase separations of an intact mAb protein (10). Whereas at 40 °C the protein peak is barely detectable, it becomes more apparent at 50 °C and keeps increasing in size up to 80 °C, which was the maximum temperature explored in this work. Based on these data and other results from our own work (11), as well as those from other groups (4), we generally use column temperatures of 70–90 °C for reversedâphase separations of proteins in our laboratories. However, when working at these temperatures, we also try to limit on-column times (that is, retention times) to less than about 20 min to minimize the likelihood that proteins will degrade inside of the column. Very recent results suggest a future where this requirement of elevated column temperature may be relaxed to some extent through development of new stationary-phase materials (7).

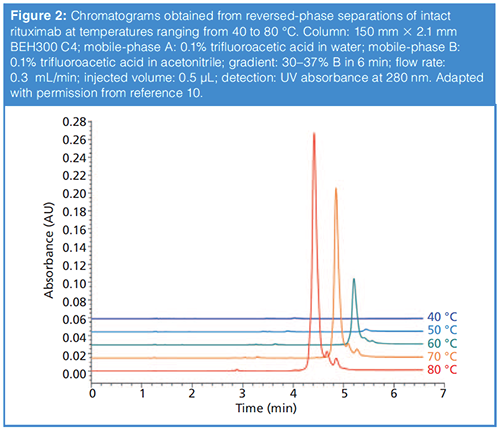
To some extent these issues of incomplete recovery (adsorption) and on-column degradation of the protein are protein- and columnâspecific. This situation means that, while we use the guidelines described above as a starting point in method development, it is also a good idea to experimentally determine the conditions needed to maximize recovery and minimize degradation for the protein or sample of interest. Realistically, the inherently complex and dynamic nature of protein structures limits our ability to predict interactions between proteins and their surroundings, and thus their adsorption and retention properties under reversed-phase conditions. For these reasons, screening methods have become quite popular as a means to empirically evaluate some specific protein properties, including chromatographic behaviour and developability as therapeutic molecules.
What About the StationaryâPhase Chemistry?
Many stationary phases marketed for reversed-phase protein separations are commercially available. Some of these are based on particles composed entirely of organic polymers. Others are based on silica and other metal oxides that have stationary phases either covalently bonded to or coated on the particle surface. As a practical matter, it is reasonable to ask how the stationaryâphase chemistry affects reversed-phase separations of proteins. In the case of silica-based phases, the type of alkylsilane bonded to the surface can influence the retention of proteins and can therefore be used to manipulate the retention and, to a lesser extent, selectivity. Although the detailed molecular basis of the effect of stationary-phase ligand structure on protein retention is not fully understood, we know from experiments that a number of factors influence retention. These factors include the relative hydrophobicity of the ligand, surface coverage, ligand density, carbon load, flexibility of the ligand, and degree of exposure of the surface silanols. In addition, the choice of ligand can also influence the recovery and conformational integrity of the protein analytes.
Historically, it was assumed that shorter and less hydrophobic n-butyl ligands provide better recovery than octyl or octadecyl ligands (C8 and C18 phases, respectively). Therefore, in the past mostly butyl (that is, C4) and propyl (C3) phases were used for protein separations. However, with modern commercially available phases, it now seems that there is no significant dependence of protein recovery on the length of the stationary phase alkyl ligand (10). Significant differences in the retention of proteins are sometimes observed when comparing columns with different bonded phases. For example, in several cases, it has been observed that proteins are more retained on C4 stationary phases than they are on C8 or C18 phases. The reason for this behaviour may be that large solutes (proteins) do not penetrate into the bonded-phase layer like small molecules do (12), because the proteins are simply too big compared to the available space between ligands. In this way, proteins probably interact with the stationary phase mainly at the interface between the bonded-phase layer and the bulk mobile phase-in other words, proteins only experience a “bird’s-eye view” of the stationary phase (1). In most cases, the ligand density of shorter chains is larger than that for longer chains, thus the accessible hydrophobic surface area is larger for phases modified with short alkyl ligands compared to longer ligands. In addition, if there are residual unbonded silanols present on the silica surface, they will be more accessible in cases where the stationary phase is composed of short-chain ligands (hydrogen-bonding and ionâexchange interactions have longer interaction distances than dispersive interactions).
Our perspective is that most alkyl bonded phases (for example, C3, C4, C8, and C18) are viable options for proteins separations, provided that conditions are optimized for a particular material. This optimization should consider-at a minimum-pore size, mobile-phase temperature, and mobile-phase additives (for example, trifluoroacetic acid). Some recently introduced materials look promising from the point of view of ease of use (7), and it will be interesting to see how these offerings continue to evolve in the near future.
Summary
Over the past decade, many new materials and columns have been introduced for reversed-phase separations of proteins. The good news from this trend is that users now have many more commercially available materials to choose from. The challenge, though, is sorting out which one will be best for a particular application, as well as finding optimal separation conditions. In this instalment, we have briefly discussed the importance of pore size, column temperature, and stationaryâphase chemistry for reversed-phase separations of proteins. The ideas discussed here should be helpful to users beginning method development, or troubleshooting the performance of an existing method. Much remains to be discussed, both in terms of additional considerations for reversedâphase separations, and for other separation modes including ionâexchange and size-based separations, and we look forward to discussing these issues in future instalments.
References
- J.W. Dolan, LCGC Europe 27(4), 190–194 (2014).
- D. Bell, LCGC Europe 31(4), 202–211 (2018).
- S. Fekete, R. Berky, J. Fekete, J.-L. Veuthey, and D. Guillarme, J. Chromatogr. A 1252, 90–103 (2012). doi:10.1016/j.chroma.2012.06.066.
- S.A. Schuster, B.M. Wagner, B.E. Boyes, and J.J. Kirkland, J. Chromatogr. A 1315, 118–126 (2013). doi:10.1016/j.chroma.2013.09.054.
- B.M. Wagner, S.A. Schuster, B.E. Boyes, T.J. Shields, W.L. Miles, M.J. Haynes, R.E. Moran, J.J. Kirkland, and M.R. Schure, J. Chromatogr. A 1489, 75–85 (2017). doi:10.1016/j.chroma.2017.01.082.
- W. Chen, K. Jiang, A. Mack, B. Sachok, X. Zhu, W.E. Barber, and X. Wang, J. Chromatogr. A 1414, 147–157 (2015). doi:10.1016/j.chroma.2015.08.043.
- B. Bobály, M. Lauber, A. Beck, D. Guillarme, and S. Fekete, J. Chromatogr. A 1549, 63–76 (2018). doi:10.1016/j.chroma.2018.03.043.
- H. Chen and C. Horvath, Anal. Methods 1, 213–222 (1994).
- B.C. Trammell, L. Ma, H. Luo, M.A. Hillmyer, and P.W. Carr, J. Am. Chem. Soc. 125, 10504–10505 (2003). doi:10.1021/ja036339d.
- S. Fekete, S. Rudaz, J.-L. Veuthey, and D. Guillarme, J. Sep. Sci. 35, 3113–3123 (2012). doi:10.1002/jssc.201200297.
- S. Fekete, A. Beck, E. Wagner, K. Vuignier, and D. Guillarme, J. Sep. Sci. 38, 1–8 (2015). doi:10.1002/jssc.201400996.
- M.R. Schure, J.L. Rafferty, J.I. Siepmann, and L. Zhang, LCGC Europe 27(1), 18–27 (2013).
Szabolcs Fekete holds a Ph.D. degree in analytical chemistry from the Technical University of Budapest, Hungary. He worked at the Chemical Works of Gedeon Richter Plc at the analytical R&D department for 10 years. Since 2011, he has worked at the University of Geneva in Switzerland. He has contributed to more than 100 journal articles and authored book chapters and edited handbooks. His main interests include liquid chromatography (reversed phase, IEX, SEC, HIC, SFC, HILIC), column technology, method development, and pharmaceutical and protein analysis.
Davy Guillarme holds a Ph.D. degree in analytical chemistry from the University of Lyon, France. He is now senior lecturer at the University of Geneva in Switzerland. He has authored more than 200 journal articles related to pharmaceutical analysis. His expertise includes HPLC, UHPLC, HILIC, LC−MS, SFC, and analysis of proteins and mAbs. He is an associate editor of Journal of Chromatography B and editorial advisory board member of several journals including LCGC North America, Journal of Chromatography A, Journal of Separation Science, and others.
Dwight R. Stoll is the editor of “LC Troubleshooting”. Stoll is a professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota, USA. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 50 peerâreviewed publications and three book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: LCGCedit@ubm.com













Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



