The LCGC Blog: Water Immiscible Solvents as Sample Diluents in Reversed-Phase HPLC – You Must be Joking!
Many of us have faced the situation where we have analytes that vary widely in their polarity or LogP(D) values and encounter issues with analyte solubility when choosing a suitable sample diluent for our high-pressure liquid chromatography (HPLC) analysis. The more polar analyte will favor aqueous solvents, and the less polar will be more highly soluble in organic solvent—so which do we choose?
We often plump for an intermediate solvent such as 50:50 Water:acetontrile (or methanol) to make the diluent as favorable as possible to ensure all analytes as fully dissolved as possible prior to analysis. However, this is often where we run into problems with peak shape deformation of early eluting peaks especially when gradient HPLC is employed. There is also the possibility that neither acetonitrile nor methanol are able to solubilize the hydrophobic analytes of interest. Further, we may have an organic eluent from a sample preparation technique that we wish to inject directly into our HPLC without risking drying down and re-constituting in an aqueous-based diluent (solvent exchange). What are our options under these circumstances?
Well, contrary to popular belief, it is possible to use water immiscible solvents or solvent combinations as injection solvents when the solubility of analytes is limited or if we wish to introduce an organic extract directly into the HPLC system, however there are a few basic rules that we need to follow for successful analysis.
So, let’s start by examining the so called “general elution” problem, where we need to dissolve our analytes in a diluent that contains a higher proportion of organic solvent than the mobile phase at the start of the gradient.


Figure 1: Separation of two model analytes using reversed-phase HPLC: C18 100 x 2.1 mm, starting gradient composition 90:10 aq: MeCN for 1 min followed by linear gradient to 70% MeCN in 12 min: Column representations: Green – analyte band / Blue – weaker solvent: sample diluent 90:10 aq: MeCN: injection volume 10 mL.
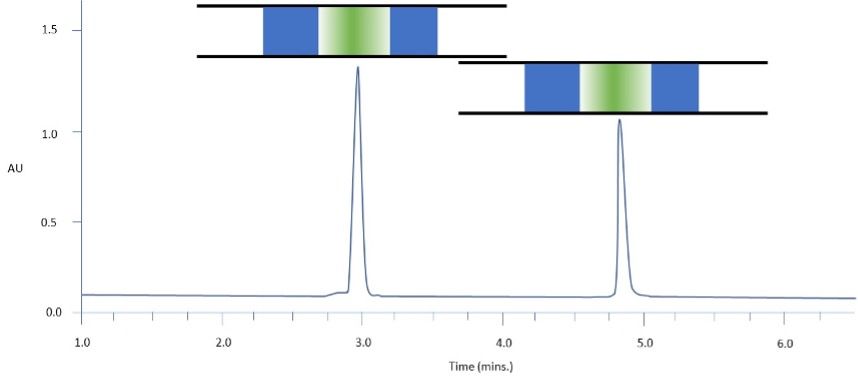
Figure 1 shows two model analytes during “ideal” gradient analysis circumstances. Here the diluent has the same or lower elution strength than the starting composition of the eluent in a gradient analysis. For example, if the eluent composition at the start of the gradient is 90:10 water: acetonitrile, then the diluent will contain 10% or less acetonitrile by volume. As the sample plug is injected into the HPLC system and enters the column, and the sample diluent and eluent mix, then the elution strength of the solvent environment for each analyte will be approximately equal. The analyte will travel through the column and there will be no solvent-based driving force for peak broadening or splitting, and this is the ideal situation for gradient HPLC analysis. For clarity, all of the other intrinsic band-broadening processes associated with HPLC will still be in play, however there will be no further, solvent-based, band broadening.
As I’ve explained above, there are several circumstances in which this ideal situation cannot be achieved, and often we need to increase the “elution strength” of the sample diluent above the composition of the starting gradient. This situation risks both peak broadening and deformation, especially for earlier eluting analytes.

Figure 2: Separation of two model analytes using reversed phase HPLC; C18 100 x 2.1 mm, starting gradient composition 90:10 aq: MeCN for 1 min followed by linear gradient to 70% MeCN in 12 min; Column representations: Green – analyte band / Blue – weaker solvent / Red – stronger solvent; sample diluent 40:60 aq: MeCN; injection volume 10 mL.
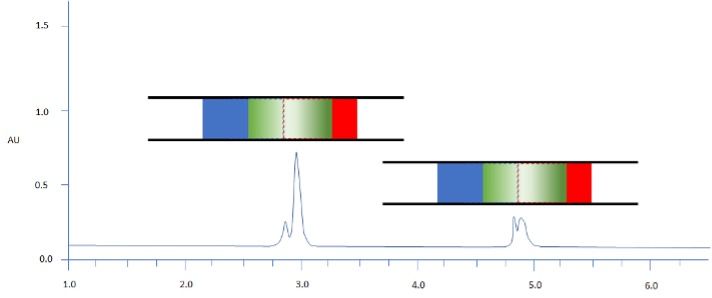
Figure 2 shows why this might be the case. The red solvent is the more highly eluting sample diluent, and the blue is the mobile phase, which has a lower elution strength. As the injected sample mixes at the head of the analytical column, the analytes are distributed across a zone which has markedly differently elution strength characteristics, and so some analytes travel down the column faster and some will travel slower. The chromatogram in Figure 2 highlights the issues with peak broadening and splitting due to the “differential” sample elution speed of analytes in either the faster or slower eluting solvents. These peak shapes are far from ideal and will certainly give us problems with peak integration and destroy the faith any reviewer of the data has in our abilities as chromatographers!
There are several precautions that we can take to reduce these effects including:
- Using the lowest amount of organic solvent within the diluent that will produce the required solubility characteristics for the analytes of interest
- Using the lowest injection volume possible to ensure the optimum mixing of eluent and diluent PRIOR to the sample reaching the head of the analytical column (for example, within the tubing, eluent pre-heating chambers, and connections between the autosampler and the analytical column)
- Using a low-volume inline filter at the outlet of the injection valve to promote the mixing of the diluent and eluent, again achieving a more homogeneous solvent plug prior to arrival at the head of the analytical column
However, what if we need to revert to an alternative organic solvent as the sample diluent for solubility reasons, or if we wish to avoid a solvent-exchange step from our sample preparation eluate? Well, it is possible to use much less conventional, even water immiscible, solvents as diluents if we bear one primary rule in mind, and that is, the solvent we use must elute later than our analytes of interest or, if this is not possible, then much earlier than them.

Figure 3: Separation of two model analytes using reversed-phase HPLC; C18 100 x 2.1 mm, starting gradient composition 90:10 aq: MeCN for 1 min followed by linear gradient to 70% MeCN in 12 min; Column representations: Green – analyte band / Blue – weaker solvent / Red – stronger solvent; sample diluent ethyl acetate; injection volume 10 mL; S = sample solvent peak
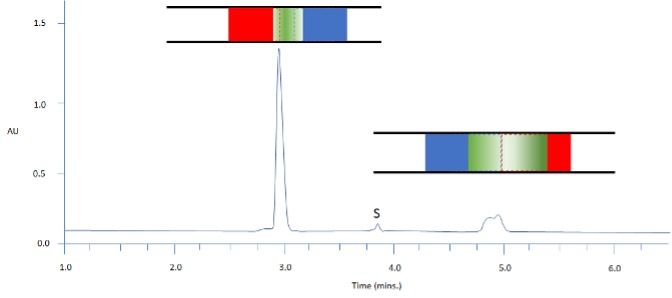
Figure 3 demonstrates this principle with our model analytes, unfortunately, we can see that a solvent has been chosen which elutes between the two analytes and whilst the earlier eluting analyte has regained its peak shape, the analyte eluting later than the solvent front gives us a very poor peak shape. Again, we can rationalise this behavior in terms of the distribution of analyte between the two solvent regions. Where the organic solvent, ethyl acetate in this case, elutes more slowly than the analyte, there is no driving force for the analyte band to broaden as the font of analyte band is not moving faster than the back of the analyte band. Whilst the organic solvent still has a higher elution strength, as long as it does not carry the front of the analyte band with it, then no broadening will occur. Conversely, for the later-eluting analyte, we are back to same position as we had in Figure 2, where the front of the analyte band is carried significantly further down the column than the back of the band and peak deformation occurs.

Figure 4: Separation of two model analytes using reversed-phase HPLC; C18 100 x 2.1 mm, starting gradient composition 90:10 aq: MeCN for 1 min followed by linear gradient to 70% MeCN in 12 min; Column representations: Green – analyte band / Blue – weaker solvent / Red – stronger solvent; sample diluent MIBK; injection volume 10mL; S = sample solvent peak.
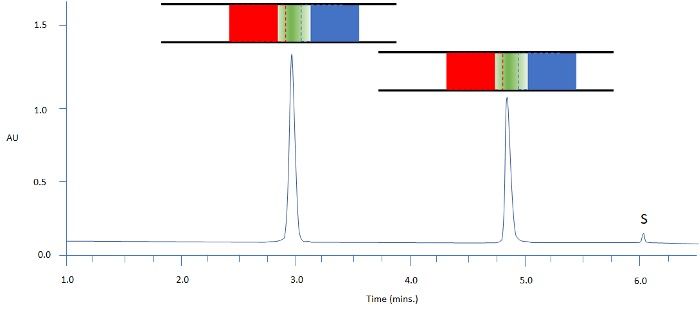
In Figure 4, we have chosen methyl isobutyl ketone (MIBK) as the sample diluent, which elutes later than both of the model analytes. Here we achieve the ideal situation in which both analyte bands encounter weaker mobile-phase strength at the front of the band and a more strongly eluting mobile phase at the rear of the analyte band as the sample enters the column. As described earlier, neither analyte experiences a driving force for severe peak broadening or splitting, and one could strongly argue that these peak shapes would be acceptable for integration and save our reputation as decent chromatographers!
We should be realistic here and comment that in any situation where our analytes experience differential elution strength environments as they enter the HPLC column, there is a risk of a small degree of band broadening, and this risk increases as the volume of the sample injection increases. As such, we should still seek to limit the volume of sample injected to minimize any peak broadening.
Many different organic solvents can be used in this manner, the important point being the relative elution time between the diluent solvent and your analytes, and this may need a little trial and error to optimize. I have had success with iso-propanol, ethyl acetate, MIBK and toluene to name but a few diluent solvents. It is also possible to mix small amounts of acetonitrile with these solvents, in various proportions, depending upon the required solubility characteristics required for your sample mixture.
So, the next time you are considering moving to hydrophilic interaction chromatography (HILIC) using a solvent-exchange step for your organic eluate from your sample preparation step or, even more radically, thinking of moving to normal-phase chromatography, ask yourself if using an organic solvent as your diluent might just solve your problem with a great deal less fuss!


Tony Taylor is the Chief Scientific Officer of Arch Sciences Group and the Technical Director of CHROMacademy. His background is in pharmaceutical R&D and polymer chemistry, but he has spent the past 20 years in training and consulting, working with Crawford Scientific Group clients to ensure they attain the very best analytical science possible. He has trained and consulted with thousands of analytical chemists globally and is passionate about professional development in separation science, developing CHROMacademy as a means to provide high-quality online education to analytical chemists. His current research interests include HPLC column selectivity codification, advanced automated sample preparation, and LC–MS and GC–MS for materials characterization, especially in the field of extractables and leachables analysis.


. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")


New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).


