The LCGC Blog: Inexpensive, Quick, and Selective: Seeking the Holy Grail of Sample Extraction
Oliver Napoleon Hill (1883– 1970) was an American self-help author once described as ”the most famous conman you’ve probably never heard of” (1 ). Conman maybe, but there is a quote of his that I believe to be particularly true when considering sample preparation for chromatography techniques; ”The one who tries to get something for nothing generally winds up getting nothing for something.”
I have many conversations with colleagues and customers who want to ”keep analyte extraction simple,” to “make sure the extraction method is rapid and robust,” to ”ensure the extraction method is cost-effective, as we have so many samples to process.” All very noble aims.
Except, these same people will tell me that limits of detection aren’t looking great, specificity isn’t too good, there are some coelution issues, or that they are having problems with matrix effects in LC-MS. I refer you to the learned gentleman above, and add that to achieve quick, cheap, and good all at once takes a lot of work in development. Oh, I nearly forgot: that’s the other thing that I hear a lot these days, ”we don’t have a lot of time to get this method up and running!” Exasperation.
All this being said, depending upon your analytical goals, there are some techniques, which when properly optimized, can achieve that holy grail of inexpensive, quick, and selective. But in order to efficiently develop these methods, one needs to know the premise on which they work and how to quickly optimize them. We begin this short series by describing the principles and optimization of perhaps the most straightforward of all the extraction techniques, liquid-liquid extraction (LLE).
No matter which ”version” of liquid-liquid extraction method you wish to use, there are some fundamentals that apply, and perhaps principle among these is analyte physicochemical information. It can be shown that, for neutral molecules, the partition coefficient between water and an organic solvent is directly related to the analyte hydrogen bond acidity, hydrogen bond basicity, dipolarity, polarizability, and molar volume (2). For ionizable species, at least one further descriptor related to the properties of the ionic form is required. It is, however, my experience that predictive relationships of this kind are rarely used in practice, and a trial and error approach using various solvent combinations is typically undertaken.
Understanding analyte physico-chemical parameters
To achieve the best selectivity and recovery, one needs to ensure that partitioning of target analytes from one phase into the other is as efficient as possible, and this is underpinned in most cases by some simple physicochemical information. The following information is recommended;
- LogP (D) (essential)
- pKa (essential for ionizable compounds)
- Number of hydrogen bond donors or acceptors (useful)
- Specific volume (inverse of density) (m3/Kg) (useful)
For example, this data for the molecule Cathinone, which is an ionogenic substance as shown in Figure 1:


Figure 1: Dynamic equilibrium for the target analyte Cathinone
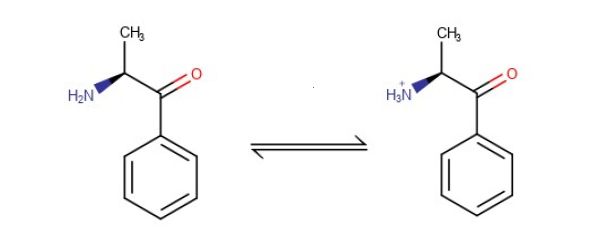
- LogD (pH10) = 1.18
- Log D (pH2) = -1.86
- pKa = 7.55
- Number hydrogen bond donors 1
- Specific Volume 0.91
This information is available from a variety of sources and we have made extensive use of the following;
- Chemspider (Royal Society of Chemistry, UK) (http://www.chemspider.com/)
- Chemicalize (ChemAxon) (https://chemicalize.com/)
- UFZ-LSER database (https://www.ufz.de/) (see also Ref. 4)
- Marvin Sketch (ChemAxon) (which also calculates values for analytes not in the previous databases) (https://chemaxon.com/products/marvin)
So how can use this information to design and optimize our LLE experiment. The first choice in designing any liquid extraction is the choice of extracting solvent, and this choice is driven by the relative hydrophobicity of the analyte molecule, which is reflected by its LogP(D) value.
As a brief reminder, the partitioning behaviour of an analyte between two phases will be reflected in LogP values as follows:

Table I: Analyte phase distribution for different LogP values
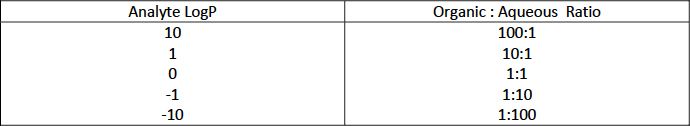
As can be seen from Table I, the analyte Log P value will indicate the degree of success that is likely in extracting an analyte from an aqueous matrix.
Analyte pH manipulation
It is obvious from the Cathinone physico-chemical data that the LogP(D) value will vary according to the pH of the sample solution, and we can use this information to our advantage when designing LLE experiments. For our Cathinone example, by adjusting the aqueous pH to a higher value, the analyte will be in the neutral form and therefore will be to the left of the equilibrium and the analyte LogD value (as the analyte is ionizable we used LogD rather than LogP but the general relationships hold) will be 1.18 and will partition 10:1 into the organic phase, depending upon the organic solvent chosen.
The relationship between solution pH and pK is shown below as a reminder for the target pH values:


For our target Cathinone analyte, adjusting the sample solution pH to 9.55 or above, will ensure the analyte is in the neutral form, the LogD value will be 1.18, and therefore will be more successfully extracted from the aqueous sample into an organic solvent. This should demonstrate the importance of knowing the analyte physicochemical properties in designing your extraction experiment. If the sample solution had been at neutral or acidic pH the partition coefficient for the analyte would have been much lower and recovery from the sample solution into the extracting solvent would be accordingly very much lower.
If one is unsure of analyte pKa or even exact analyte structure, it is always best practice to assess the effect of pH on solvent extraction efficiency with the following pH conditions suggested;
- pH 9 and 11 for suspected basic analytes
- pH 6 and 7.5 for suspected neutral analytes
- pH 2.5 and 4 suspected acidic analytes
The difference in analyte recovery between each of the pH value pairs can sometimes be quite startling and these differences are driven by the (unknown) analyte pKa.
Back extraction
The selectivity of the extraction experiment involving pH manipulation can be further improved using a technique known as back extraction. Once the target analytes are in the organic phase, they can be re-extracted into a fresh aqueous phase whose pH has been manipulated to ensure the analytes are in the charged form and therefore most highly hydrophilic. For acidic target analytes, the second aqueous extracting solution is manipulated to basic pH and vice versa for basic analytes. In this way, specificity of the extraction can be improved as all neutral extractants will be left behind in the organic solvent, reducing the instances of co-elution, and achieving lower matrix background (and hence lower detection limits). There are no limits to the number of back extraction steps that can be employed to improve specificity and hence the aqueous layer pH may again be adjusted to ensure the analyte is in the neutral form and extracted into a further organic solvent. Of course, absolute analyte concentration will decrease in each back-extraction stage according to the specific recovery achieved at each stage.
Use of salts
Recovery of more highly hydrophilic analytes from aqueous samples can be promoted through the use of salt addition to the sample. There are two common approaches:
- Use of an ion-pairing salt with opposite charge to the analyte, which, if the sample solution pH is adjusted to ensure the analyte is fully ionized, will pair with the analyte to form a neutral complex. This neutral complex is likely to have a much higher partition co-efficient than the ionized analyte, and recovery into the organic extracting solution will be increased. When analytes are strongly acidic or basic, this tends to be the method of choice.
- Adding high-enough concentrations of simple salts to saturate the aqueous sample solution can increase the partition coefficient of hydrophilic analytes and improve recovery into organic extraction solvents. Effectively the analyte solubility in the aqueous sample is reduced by adding, for example, 3–5 M sodium sulphate to the sample solution, driving the target analytes into the organic phase and improving recovery. There are a variety of salts that can be used in this way, and the required concentrations to achieve saturation, or optimum analyte recovery, need to be empirically determined.
Extraction solvent choice
Of course, the partitioning behavior of our target analytes is strongly influenced by the choice of organic solvent into which we chose to extract our analyte, as the various solvents will also have a range of polarities that will influence their affinity for the analyte(s). For reference, some water-immiscible solvents are shown below with their polarity index values.

Table II: Polarity index values of a variety of popular organic solvents use for LLE
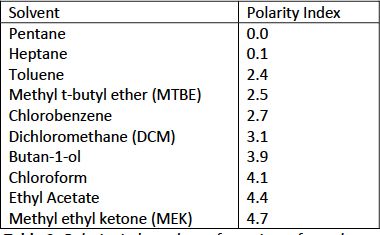
For more polar analytes, and here lower values of LogP(D) can also act as a good indicator of polarity, one may need to select an organic solvent that has a higher polarity index value in order to optimize recovery of the analyte from the aqueous sample into the organic extracting solvent. The rules are fairly simple: try to match the polarity of your analyte with the polarity of the extraction solvent. If you cannot obtain a good polarity measure of the analyte (see later discussion for some useful tools to help with this), LogP(D) can be used as a surrogate, with lower LogP values indicating the need for a more polar extraction solvent and vice versa.
Of course, it is also possible to used ”mixed” organic systems as the extraction solvent to further fine-tune the selectivity or the recovery of the target analytes from the sample. Here one needs to develop an in-house screening approach, using solvent mixtures that have proven to be successful in your application area. To assist with this, a list of the solvent systems I have found particularly useful in the past is shown below in Table III. We typically use 96 well plates to screen these solvent mixtures, as this provides a rapid way of assessing the gross partitioning behaviors of a relatively large number of analytes, several solvents, and perhaps different pH or (as we shall see later), and salt strengths and still leaves enough wells for blanks, recovery standards, and so on.

Table Table III: Useful solvent mixtures for fine tuning extraction recovery in LLE
Of course, the most effective solvent combination in terms of recovery will depend upon the nature of the analyte and interferent species that we wish to leave behind, and some trial and error is likely.
Advanced methods for extraction solvent selection
I recently came across a fascinating paper in which the group had investigated building a predictive algorithm for extraction efficiency based on quantitative relationships between the LogP(D) values of model compounds in a variety of solvent pairs. (2) It strikes me that this would be a very logical and useful approach where one is working in a specific application area. A suitable set of model compounds can be identified. This group used pKa, Log D, number of hydrogen bond donor (HBD) groups, and the analyte specific volume to calculate the analyte partition coefficient in a variety of organic solvents from an equation derived from only a few basic partition co-efficient measurements.
There are other online tools that can help to inform the optimum choice of solvent for liquid-liquid extraction and perhaps the most useful of these is the UFZ-LSER database (3). Using the SMILES string for our cathinone molecule, it was possible to calculate some key thermodynamic and physico-chemical properties of the molecule that are then used to predict the partition coefficients between a wide variety or organic solvents (solvent mixtures) and water. This is a very useful tool to quickly identify the organic solvents to use in any screening experiments and, in this particular example, it was identified that chloroform is a good candidate for the liquid extraction of Cathinone. While the results from this database are less reliable when the analyte properties need to be calculated, it does contain a vast array of compounds with known values, and will also search the internet for reliable sources of the descriptors, again improving the reliability of the partition coefficient estimations.
A further cautionary note when selecting the extraction solvent is that one always needs to be mindful of which layer is the extraction solvent, bearing in mind that certain organic solvents are more dense than water and will therefore be the lower layer in the extraction vessel. Common examples of these solvents include (in order of increasing density):
Chlorobenzene < Ethylene Dichloride < o-Dichlorobenzene < Methylene Chloride < Trichloroethylene < Chloroform < Carbon Tetrachloride
Improving selectivity of LLE
When considering solvent selection, one must always be mindful of the specificity of the extraction, and here I specifically refer to the ability of technique to separate the target analytes from matrix components which may have similar chemical and physico-chemical properties. Where simple binary solvent systems are used for liquid-liquid extraction, the control of specificity can be restricted to the choice of the organic solvent, the sample solution pH, and perhaps the ionic strength. In terms of organic solvent selection, one important selectivity consideration is that of the water solubility in the organic solvent, and this can be particularly important when one is carrying out trace analysis. The relative solubility of water in a range of organic solvents is shown in Table IV:

Table IV: Water solubility for various organic solvents popular in LLE
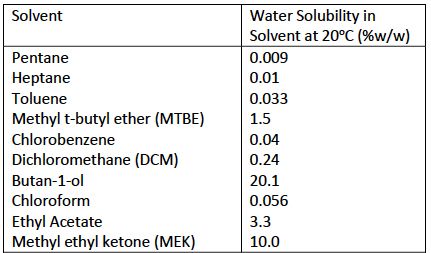
From Table IV, it should be obvious that some organic solvents are able to solubilize a reasonable amount of water and therefore can be expected to be less specific than those solvents in which water has much lower solubility. Using solvents that can more readily solubilize water will give rise to extracts which contain higher levels of interferents which, as is pointed out above, may be a particularly important considering when performing trace analysis. A very useful lookup resource for these water solubilities can be found at the following link: https://macro.lsu.edu/HowTo/solvents.htm
Extraction to sample solvent ratio
To ensure the highest recovery possible, the ratio of organic extraction solvent to aqueous sample should be high (or perhaps higher than is generally accepted as reasonable), and a ratio of 7:1 is considered somewhat of a generic optimum value. Obviously, the actual optimum value will depend upon the partition coefficient of the target analyte(s) between the two solvents, and extraction solvents with high partition coefficients may require lower ratios and vice versa. Interestingly, the database of Reference 4 has a nice tool (“Extraction Tool”) which allows the optimum solvent/sample ratio to be estimated, which can save a great deal of empirical optimization. The optimum value for the Chloroform extraction of Cathinone as described above was found to be 6:1 organic to aqueous using this tool, indicating that our generic 7:1 ratio for optimum recovery may indeed be a reasonable guide.
Volatility of extraction solvent and analyte
The volatility of the extracting solvent is a further important consideration that goes alongside the selection of the optimum organic : aqueous ratio, primarily as the speed of sample preparation will usually be rate limited by the speed with which the organic solvent volume may be reduced, using evaporation to achieve a sample concentration that meets analytical limits of detection. More volatile extraction solvents may give rise to more rapid evaporation protocol steps and therefore reduce the overall extraction experiment time, but it should be stated that if one can avoid a solvent evaporation step, then this is much preferred. Solvent evaporation steps can introduce unwanted qualitative and quantitative error into the determination, and the careful choice of solvents with high affinity for the target analytes may allow lower solvent volumes to be used and may, perhaps, avoid the need for solvent evaporation.
A further, often-overlooked consideration is that of analyte volatility. An ideal solvent should be capable of evaporation-to-dryness in a reasonably short time (10–15 min) under nitrogen when heated to 30– 40 oC. If the analyte can withstand these conditions without loss or degradation, then recovery will be high. Of course, the more volatile the analyte under these conditions, then the lower the recovery of the extraction experiment. If recovery is low due to analyte volatility, one must consider an alternative extraction solvent that will evaporate under less harsh conditions.
Mixing (agitation) mode and extraction time
The facile process of extraction requires that the surface area interface between the two phases is maximized, which will often involve the mixture being agitated with reasonable vigor. In the modern laboratory, manual shake flasks have been (or should be!) replaced with smaller vessels that can be mixed on an orbital mixer or by using automation systems. Yet one still needs to optimize the vigor with which the sample is mixed, in order to achieve maximum analyte recovery in a reasonable timeframe. Of course, the time of extraction will also need to be optmized, and this will be predicated by the mode of mixing, affinity (partition coefficient) of the extracting solution for the analyte, and the ratio of sample-to-extraction solvent. One might consider then that there are enough variables here to perform an optimization of the extraction using Design of Experiments (DoE) approaches, and this indeed is a highly effective way to achieve the ultimate extraction recovery.
In summary then, there are many more considerations when designing a simple liquid-liquid extraction experiment than one might realize. However, to avoid ”getting nothing for something,” and to drive towards fast, inexpensive, and selective LLE protocols, one really does need to put in a lot of method development effort. There are very few shortcuts, but hopefully the discussion above and the resources highlighted will assist you in this work.
Of course ”standard” liquid-liquid extraction is only one version of the liquid extraction technique. There are many variants available, including:
- Solid supported liquid-liquid extraction (SSLE / SLLE)
- Membrane assisted solvent extraction (MASE)
- Single drop microextraction (SDME)
- Dispersive liquid-liquid microextraction (DLLME / DiLLME)
- Hollow fibre liquid phase microextraction (HF-LPME)
Next time I will consider the principles of some of these techniques as well as their optimization.
- Novak, Matt. "The Untold Story of Napoleon Hill, the Greatest Self-Help Scammer of All Time". Paleofuture (Gizmodo). Retrieved August 13, 2019.
- M. H. Abraham, W.E. Acree, J. Org. Chem. 75, 1006−1015 (2010).
- S. Tshepelevitsh, K. Hernits, J Jenčo, J.M. Hawkins, K. Muteki, P. Solich, and I. Leito, ACS Omega 2, 7772−7776 (2017).
- N. Ulrich, S. Endo, T.N. Brown, N. Watanabe, G. Bronner, M.H. Abraham, K.-U Goss, UFZ-LSER database v 3.2.1 [Internet], Leipzig, Germany, Helmholtz Centre for Environmental Research-UFZ. 2017 [accessed on 12.07.2020]. Available from http://www.ufz.de/lserd

Tony Taylor

Tony Taylor is the Chief Scientific Officer of Arch Sciences Group and the Technical Director of CHROMacademy. His background is in pharmaceutical R&D and polymer chemistry, but he has spent the past 20 years in training and consulting, working with Crawford Scientific Group clients to ensure they attain the very best analytical science possible. He has trained and consulted with thousands of analytical chemists globally and is passionate about professional development in separation science, developing CHROMacademy as a means to provide high-quality online education to analytical chemists. His current research interests include HPLC column selectivity codification, advanced automated sample preparation, and LC–MS and GC–MS for materials characterization, especially in the field of extractables and leachables analysis.



Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

Multi-Step Preparative LC–MS Workflow for Peptide Purification
March 21st 2025This article introduces a multi-step preparative purification workflow for synthetic peptides using liquid chromatography–mass spectrometry (LC–MS). The process involves optimizing separation conditions, scaling-up, fractionating, and confirming purity and recovery, using a single LC–MS system. High purity and recovery rates for synthetic peptides such as parathormone (PTH) are achieved. The method allows efficient purification and accurate confirmation of peptide synthesis and is suitable for handling complex preparative purification tasks.



