Stability and Recovery Influences of Benzo[a]pyrene, Benzo[a]anthracene, Benzo[b]fluoranthene, and Chrysene during Sample Preparation of Plant Matrices
Quantification of European Union (EU)-priority polycyclic aromatic hydrocarbons (PAHs) in plant matrices is a crucial task. Various methods for enrichment and preconcentration, such as the preloaded-pipette tip solid‑phase extraction (SPE) (1), are available. Nevertheless, analyte recovery as a result of homogenization, sample preparation, and extraction are rarely discussed in the field of phytopharmacy. This study deals with the recovery in dry plant extracts, which are typically used in phytopharmaceuticals and reflect the actual polycyclic aromatic hydrocarbon content in the commercially available end product (2). The aim of this study was to monitor benzo[a]pyrene, benzo[a]anthracene, chrysene, and benzo[b]fluoranthene loss of spiked samples as a result of commonly-used sample pretreatment, extraction, filtering, and evaporating techniques in 1:1 (v/v) cyclohexane–ethyl acetate primulae flos and sambuci flos dry extracts. Results showed that improper sample preparation can lead to false results. In the case of benzo[a]pyrene with a deviation of 155% from the theoretical true value.



Polycyclic aromatic hydrocarbons (PAHs) are small nonpolar compounds, consisting of multiple aromatic rings deriving from pyrogenic (3), petrogenic (4), and biological origin (5). Pyrogenic PAHs are formed by a variety of combustion processes under low oxygen conditions. Sources of pyrogenic PAHs are destructive distillation of coal into coke and coal tar, thermal cracking of petroleum residuals into lighter hydrocarbons, incomplete combustion of motor fuels in cars and trucks, and incomplete combustion of wood in forest fires. Pyrogenic PAHs are generally found at higher concentrations in urban areas and in locations close to major sources of PAHs.
Petrogenic PAHs occur in crude oils which are found due to widespread transportation, storage, and use of crude oil products. Major sources are oceanic and freshwater oil spills (6), underground and above ground storage tank leaks, and the accumulation of vast numbers of small releases of gasoline, motor oil and related substances associated with transportation (7). On the other hand, it is well-known that PAHs can also be produced biologically. For example, they can be synthesized by certain plants and bacteria or formed during the degradation of vegetative matter. The mode of PAHs formation can either be natural or anthropogenic (8). As a result of their importance, the European Commission introduced maximum residue values for benzo[a]pyrene and a sum parameter of benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, and chrysene in food, herbs, and dietary supplements (9). These four PAHs were chosen according to the European Food Safety Authority (EFSA) Journal (10). 9714 PAH analyses were performed in 33 food categories and the information value of PAH8 (benzo[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, indeno[1,2,3-cd]pyrene, dibenzo[a,h]anthracene, and benzo[g,h,i]perylene), PAH4 (benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, and chrysene) and PAH2 (benzo[a]pyrene and chrysene) was compared. The sum of PAH8, PAH2, and PAH4 was calculated and compared. PAH2 and PAH4 or PAH8 showed a correlation of 0.92. PAH4 and PAH8 revealed a correlation value of 0.99. Therefore, the focus of the publication was put on the subset of the PAH4, because they are required by the legislation. Screening a larger subset of PAHs could provide additional value (10).
In the near future phytopharmaceuticals are also likely to receive maximum residue values. Therefore, an accurate quantification method is mandatory, whose performance relies on an efficient extraction and a selective enrichment procedure.
Quantification of PAHs is mainly performed using high performance liquid chromatography fluorescence detector (HPLC–FLD) (11–16), or gas chromatography mass spectrometry (GC–MS) (17–19). Furthermore, liquid chromatography–atmospheric pressure photoionization–mass spectrometry (LC–APPI–MS) (20–22) techniques and also two dimensional GC (GC×GC) methods are reported (23). In terms of European Union (EU) priority, PAHs benzo[c]fluorene, benzo[a]anthracene, chrysene, 5-methylchrysene, benzo(j)fluoranthene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, dibenzo[a,l]pyrene, dibenzo[a,h]anthracene, benzo[g,h,i]perylene, indeno[1,2,3-cd]pyrene, dibenzo[a,e]pyrene, dibenzo(a,i)pyrene, dibenzo(a,h)pyrene, and cyclopenta(c,d)pyrene, HPLC–FLD methods have the disadvantage that only 15 of the 16 PAHs can be separated. This is because cyclopenta(c,d)pyrene shows no fluorescence. For the Environmental Protection Agency (EPA)-priority PAHs, naphthalene, 1-methylnaphthalene, 2-methylnaphthalene, acenaphthene, fluorene, phenanthrene, anthracene, fluoranthene, pyrene, benzo[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, dibenzo[a,h]anthracene, benzo[g,h,i]perylene, and indeno[1,2,3-c,d]pyrene, the situation is similar. Acenaphthylene cannot be resolved in HPLC–FLD due to the lack of fluorescence. GC–MS analysis can overcome this issue, but is limited by the low volatility of high-molecular‑weight PAHs. Dibenzopyrenes elute only at very high temperatures, very close to or above the thermal stability of the column, therefore decreasing the column lifetime drastically (24). Nevertheless, GC–MS has been recommended in many official methods for the analysis of PAHs (25). Liquid chromatography–electrospray ionization–mass spectrometry (LC–ESI–MS) techniques need a post‑column derivatization, such as tropylium ions or silver ions, and are therefore not very attractive (26,27). LC–APPI–MS methods are a good alternative and are mainly used for quantification of PAHs in complex matrix, using a high-resolution mass spectrometer (28–30).
PAH quantification methods include external calibration, internal calibration, and standard addition methods. For GC–MS and APPI–MS methods, mainly internal standard calibrations are used. The standard method is the use of isotopic forms of target PAHs (31,32). Isotopic forms of all the EU-priority PAHs and the EPA-priority PAHs are available. In March 2020 an automated pipette tip solid-phase extraction (SPE) method was published (1). The introduced method employs the analytical scheme described in this study, and standards are preloaded on a poly(styrene/divinylbenzene) SPE resin, therefore enabling rapid quantification without the need for standard preparation. Furthermore, the method was subjected to various tea samples, as well as contaminated primulae flos samples.
To remove interfering compounds, sample clean-up is of the utmost importance. Gel permeation chromatography (GPC) is primarily used for that (33,34), and manual and automated SPE methods are reported (1,35,36). In recent years, HPLC–FLD methods have also shown that they are capable of distinguishing alkylated PAHs from PAHs. In FDA method C-002.01, HPLC–FLD was used to quantify EPA PAHs in a complex sea food matrix. The method is able to separate alkylated PAHs from PAHs and allows an accurate quantification. The method was validated according to Level 3 Multi‑laboratory validation (MLV) (37).
Nevertheless, all these methods rely on plant sample homogenization by grinding and extraction using a highly apolar solvent composition, consisting of cyclohexane and ethylacetate or dichloromethane. Afterwards the PAHs are preconcentrated using SPE with silica gel, florisil alumina, polystyrene-divinylbenzene (38), saponification, liquid–liquid extraction or gel permeation chromatography. All of these solutions have one thing in common; a solvent change is necessary and therefore the extraction solvent needs to be evaporated. Most described methods use nitrogen evaporation or vacuum evaporation. Furthermore, many methods use filtration steps, applying polytetrafluoroethylene (PTFE) or regenerated cellulose (RC) filters. Another key fact represents the used vial material. PAHs are likely to adsorb on the surface of polypropylene falcon tubes. Therefore, a closer look on the recovery of benzo[a]pyrene, benzo[a]anthracene, chrysene, and benzo[b]fluoranthene during these steps is necessary.
Experimental
Reagents and Materials: Benzo[a]pyrene (≥96.0%), benzo[a]anthracene (99.0%), benzo[b]fluoranthene (98.0%), chrysene (98.0%), and ethyl acetate (99.8%) were purchased from Sigma Aldrich (Buchs, Switzerland). Acetonitrile (ACN) and methanol (MeOH) both in HPLC grade (Chromasolv) were purchased from Honeywell Riedel-de Haen (Seelze, Germany). Tetrahydrofuran (THF) and cyclohexane were acquired from Merck KGaA (Darmstadt, Germany). HPLC grade water was obtained from a Milli-Q water purification system by Millipore (Bedford, USA). 50 mL polypropylene centrifuge tubes with screw caps, PTFE, and Phenex-RC syringe filters (porosity 0.45 µm) were purchased from VWR International (Radnor, USA) and Phenomenex (Torrance, USA), respectively. 2 mL Injekt syringes from Braun (Melsungen, Germany) were used. Primulae flos and sambuci flos samples were provided by Kwizda Kräuterhandel GmbH (Linz, Austria).
Instrumentation: All samples and standard solutions were measured using an Agilent 1100 Series HPLC System (Santa Clara, USA) equipped with a G1321A fluorescence detector (FLD), a G1315B diode array detector (DAD), a G1316A thermostatted column compartment, a G1329A autosampler, equipped with a G1330B thermostat, and a G1311A quaternary pump with G1322A degasser. Analyses were performed using a Pinnacle II PAH analytical column (150 mm × 4.6 mm, 4-µm, Restek Corporation, Pennsylvania, USA). Mobile phase was a composition of 5% tetrahydrofuran in water (eluent A) and acetonitrile (eluent B). A gradient programme was executed using the following steps (min/% of eluent B): 0/80, 8/95, 9/80, 11/80. The flow rate was set to 1.5 mL/min and the injection volume was 10 µL. Column oven temperature was set to 30 °C and autosampler temperature was set to 4 °C. Determination of benzo[a]anthracene and chrysene was executed at an excitation wavelength of 270 nm and an emission wavelength of 390 nm. For benzo[b]fluoranthene and benzo[a]pyrene an excitation wavelength of 270 nm and an emission wavelength of 430 nm was used.


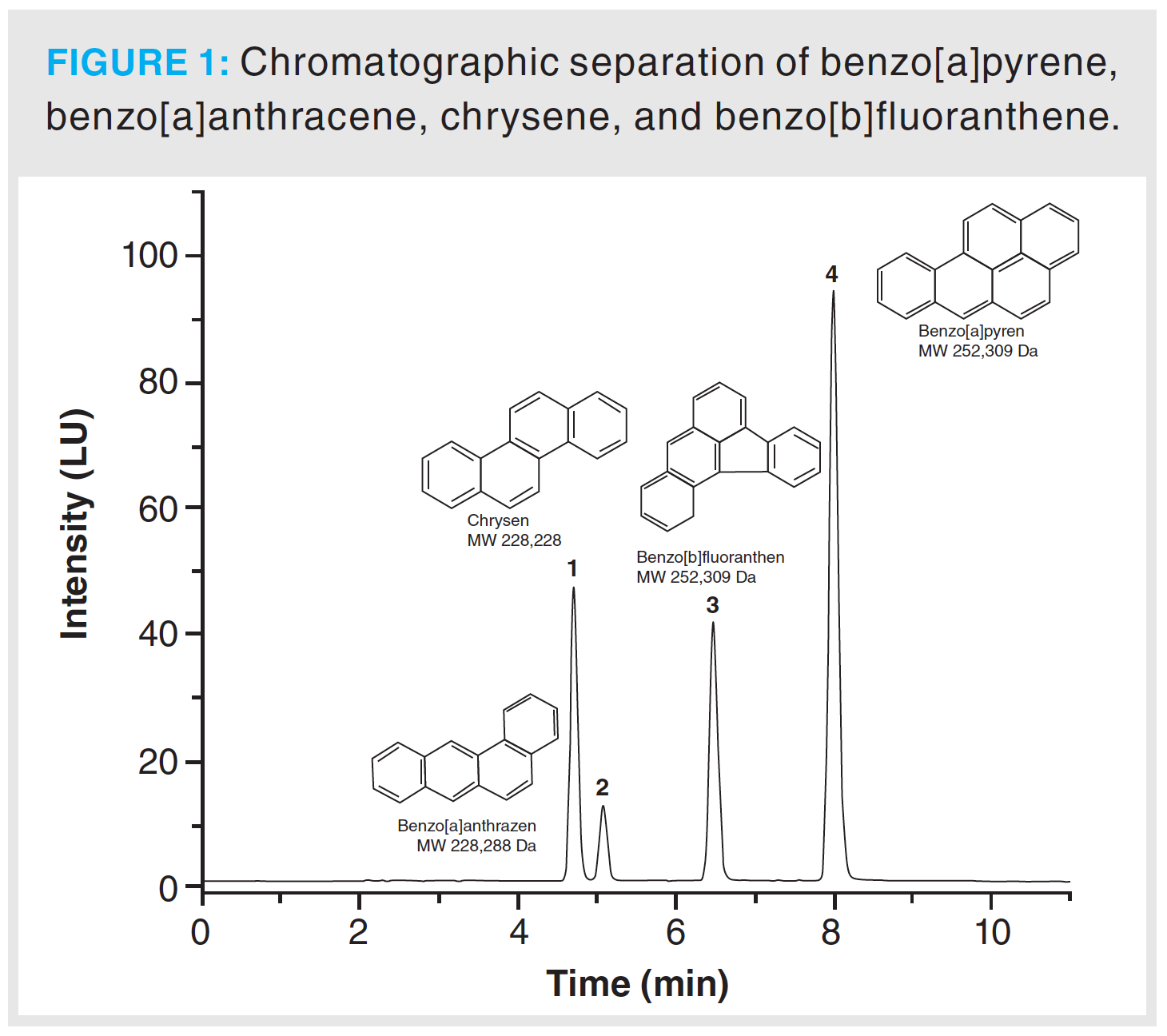
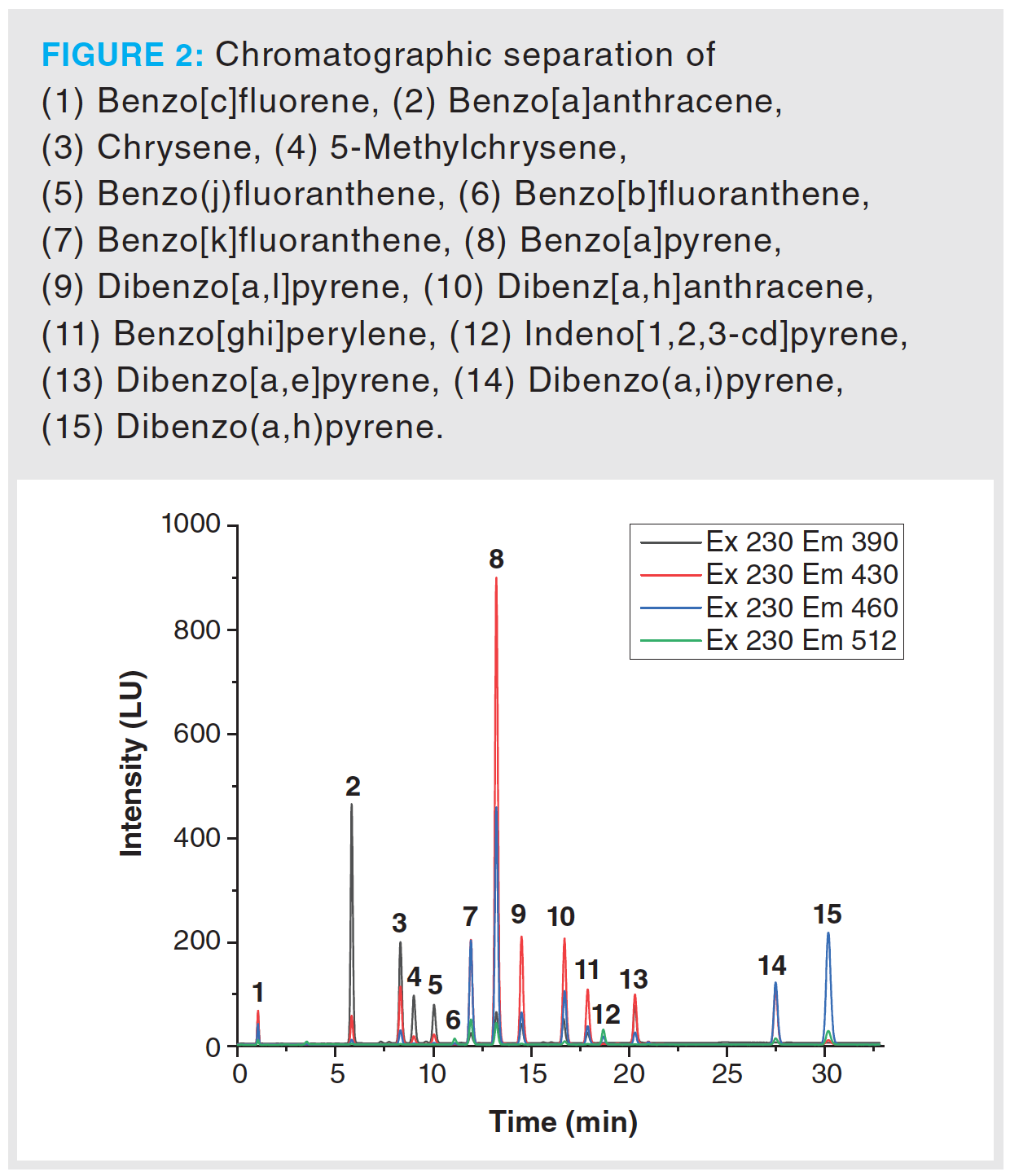
Method validation was accomplished according to international guidelines. Therefore, parameters such as accuracy, instrumental limits, linearity, repeatability, method precision, and stability of analytes were determined. Linearity was examined using a standard solution of benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, and chrysene at concentrations between 25% and 200% of target concentration (200 µg/L). For the determination of the repeatability a standard solution of 200 µg/L was prepared. Ten replicates were measured each day, to examine the intra-day repeatability. For the inter-day repeatability, three samples were measured each day for 10 consecutive days. Accuracy was investigated by spiking sambuci flos and primulae flos extracts with benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, and chrysene standard solutions at three different concentrations. Limit of detection (LOD) and limit of quantitation (LOQ) were determined from a calibration curve at concentrations ranging from 0.01 to 0.8 μg/mL. Calculations of instrumental limits were executed referring to DIN32645: 2008–11. Chromatographic separation is highlighted in Figure 1. During the method development phase, special attention was paid to avoid coelution of the EU-priority PAHs. Method development started with method 2 and the parameters were as follows: Pinnacle II PAH analytical column (4.6 × 150 mm, 4-µm, Restek Corporation, Pennsylvania, USA) was used. Mobile phase was a composition of 5% tetrahydrofuran in water (eluent A) and acetonitrile (eluent B). A gradient programme was executed using the following steps (min/% of eluent B): 0/70, 7/75, 23.5/100, 34/70, 38.7/70. The flow rate was set to 1.5 mL/min and the injection volume was 10 µL. Column oven temperature was set to 30 °C and autosampler temperature was set to 4 °C. Excitation wavelength was set to 230 nm; emission wavelengths to 390 nm, 430 nm, 460 nm and 512 nm. Figure 2 shows a measurement of the EU-priority PAHs: benzo[c]fluorene, benzo[a]anthracene, chrysene, 5-methylchrysene, benzo(j)fluoranthene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, dibenzo[a,l]pyrene, dibenzo[a,h]anthracene, benzo[g,h,i]perylene, indeno[1,2,3-cd]pyrene, dibenzo[a,e]pyrene, dibenzo(a,i)pyrene, dibenzo(a,h)pyrene, and cyclopenta(c,d)pyrene at a concentration of 800 ppb. Cyclopenta(c,d)pyrene could not be resolved as it shows no fluorescence. When optimizing the method for 15 PAHs, care was taken to ensure that no coelution of the other PAHs occurs in the final method for four PAHs.


Degradation of the employed analytes in standard solution was studied over 10 consecutive days. Therefore, standard mix solution was stored at 4 °C in the dark and the amount of each analyte was determined daily.
Plant material was homogenized using a ZM 200 centrifugal mill from Retsch (Haan, Germany) equipped with a 0.5 mm trapezoidal sieve with a 24-tooth rotor. Ultrasound-assisted extraction was performed using an Ultrasonic Cleaner USC‑TH from VWR International (Radnor, USA). Evaporation of supernatants was executed on a Concentrator plus from Eppendorf (Hamburg, Germany) or on an evaporator EVA‑VIS-72 from VLM Korrosions-Prüftechnik, Labortechnik & Dienstleistungen GmbH (Bielefeld, Germany).
Preparation of Standard Solutions: First 2 mg ± 0.10 mg of the four PAH standard substances (benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, and chrysene) were separately dissolved in 60 mL methanol in a 100 mL volumetric flasks under ultrasonic treatment. Afterwards the solution was filled up to 100 mL with methanol. These stock solutions were further used for preparation of four standard mix solutions containing all four PAH standard substances: mix 1 (800 µg/ L), mix 2 (500 µg/ L), mix 3 (200 µg/ L) and mix 4 (3000 µg/L). Therefore, the required volume of stock solution was mixed with 50 mL methanol in a 100 mL volumetric flask, homogenized and filled up with methanol to 100 mL. Working standard solutions were prepared on a weekly basis and stored at 4 °C in the dark.
Stability Evaluation of Standard Solutions: Evaluation of the stability of dissolved PAH standard substances was accomplished using the prepared standard solution mix 1, 2 and 3 (see “Preparation of Standard Solutions” section). These standard solutions were aliquoted into 1.5 mL HPLC amber glass vials (Bruckner Analysentechnik, Linz, Austria) and stored at 4 °C in the autosampler or at room temperature on a laboratory bench. Solutions were analyzed and quantified using HPLC–FLD (see “Reagents and Materials” section) over a time range of 10 days. All experiments were performed in duplicate.
Analyte Recovery After Filtration: The determination of analyte recovery after filtration was tested for PTFE and cellulose filters (see “Reagents and Materials” section) by filtering 1.5 mL of PAH standard mix 2 (see “Preparation of Standard Solutions” section) using 2 mL syringes. The permeate was collected and analyzed using the HPLC–FLD described in the “Reagents and Materials” section. Analysis was performed in duplicate.
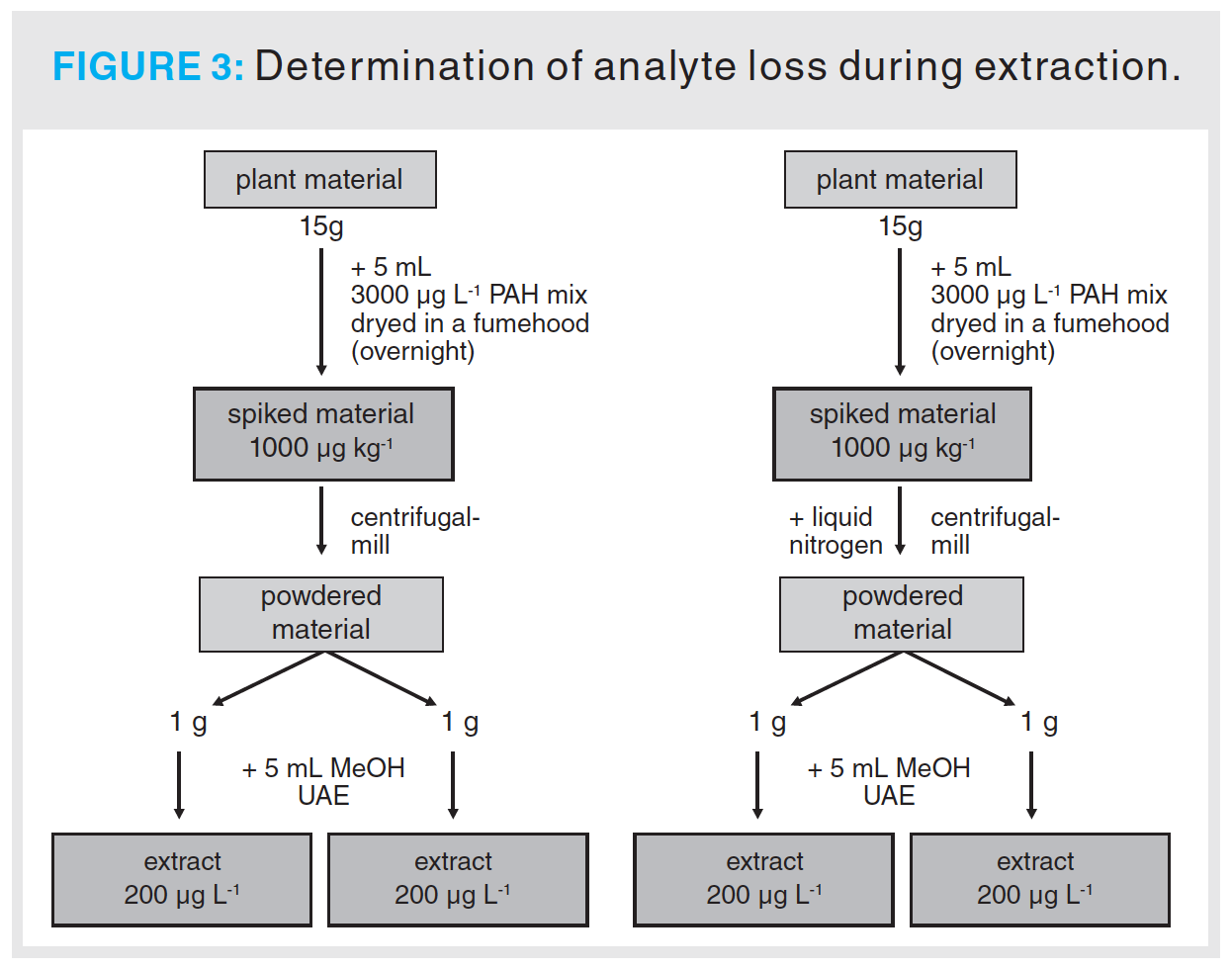
Analyte Recovery After Sample Preparation: Evaluation of the analyte recovery during sample preparation was accomplished by spiking 15 g ± 10 mg of sambuci flos plant material with 5 mL of PAH standard mix 4 (see “Preparation of Standard Solutions” section). The plant material was left for a duration of 12 h in the dark to ensure complete evaporation of the solvent. The spiked plant material was then powdered using a ZM 200 centrifugal mill from Retsch (Haan, Germany) equipped with a 24-tooth rotor and a 0.5 mm sieve at 18000 rpm. 1 g ± 1 mg of the ground plant material was extracted in 5 mL methanol using 2 min ultrasonic assisted extraction (UAE) prior to PAH quantification (see “Reagents and Materials” section). Additionally, the same procedure was performed using liquid nitrogen for cooling the samples before pulverization. The experiment was performed in duplicate. The same experimental conditions were used for primulae flos as plant material. Figure 3 shows the process sequence for the determination of the analyte recovery during sample preparation.


Analyte Recovery After Extraction: For proper extraction of PAHs from plant matrices, 1 g ± 10 mg of the powdered material was extracted in 10 mL 1:1 (v/v) cyclohexane–ethyl acetate using 30 min UAE cooled with ice, followed by centrifugation at 4500 rpm for 10 min to separate insoluble plant parts from the extraction solution. To guarantee a quantitative enrichment of PAHs using preloaded-pipette tip SPE a change of solvent to methanol was performed. Determination of analyte loss during the evaporation process was accomplished by spiking the extraction solutions of sambuci flos with 1 mL of PAH standard mix 2 (see “Preparation of Standard Solutions” section), followed by evaporation using vacuum concentration with a SpeedVac concentrator (Eppendorf, Hamburg, Germany) or nitrogen evaporation with an EVA-VIS 72 evaporator (VLM Korrosions-Pruftechnik, Labortechnik& Dienstleistungen GmbH, Biefelefld, Germany). The PAHs from the received dry extract were resolved in 1 mL methanol using 2 min UAE and quantified using HPLC–FLD (see “Instrumentation” section). The experiment was performed in duplicate. The same experimental conditions were used for determination of analyte loss as a result of different extraction vessel materials using glass eprouvettes and standard polypropylene falcon tubes.
Results and Discussion
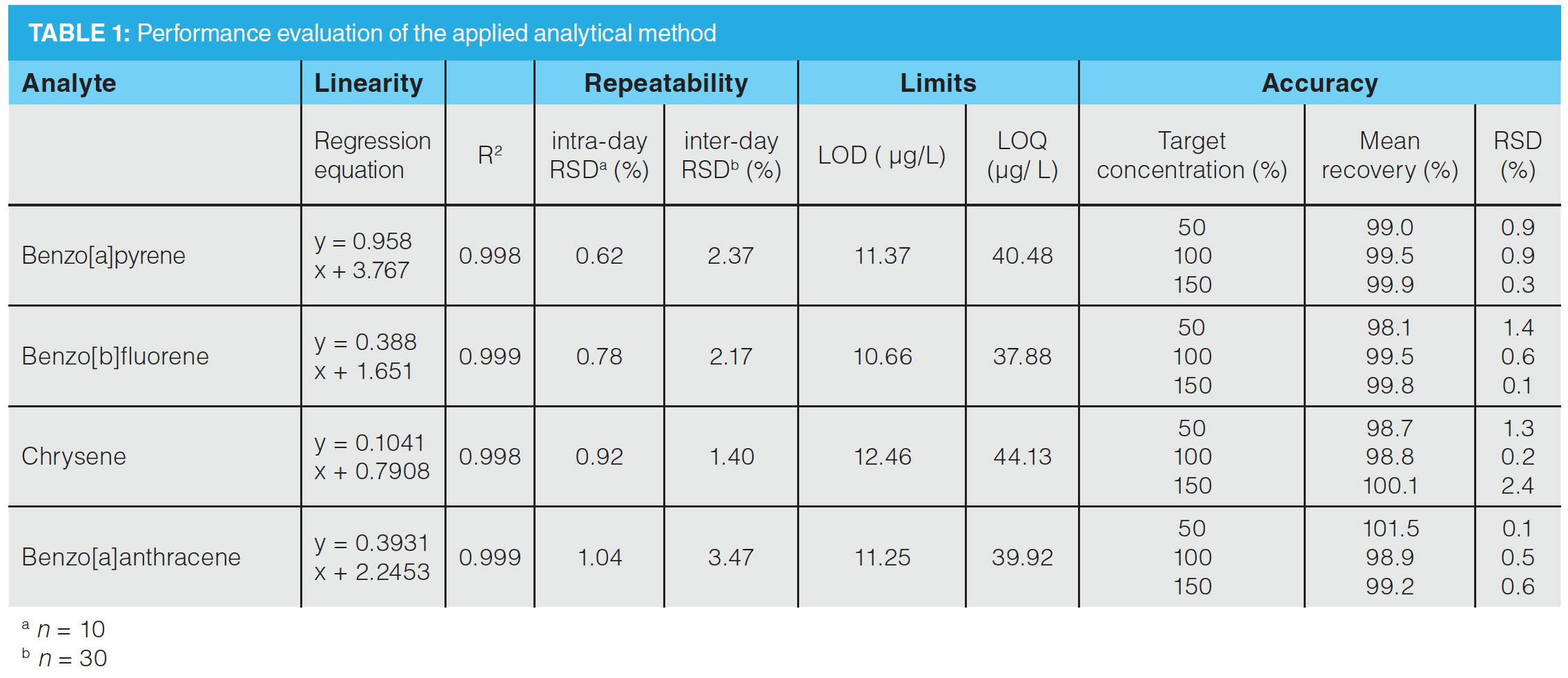
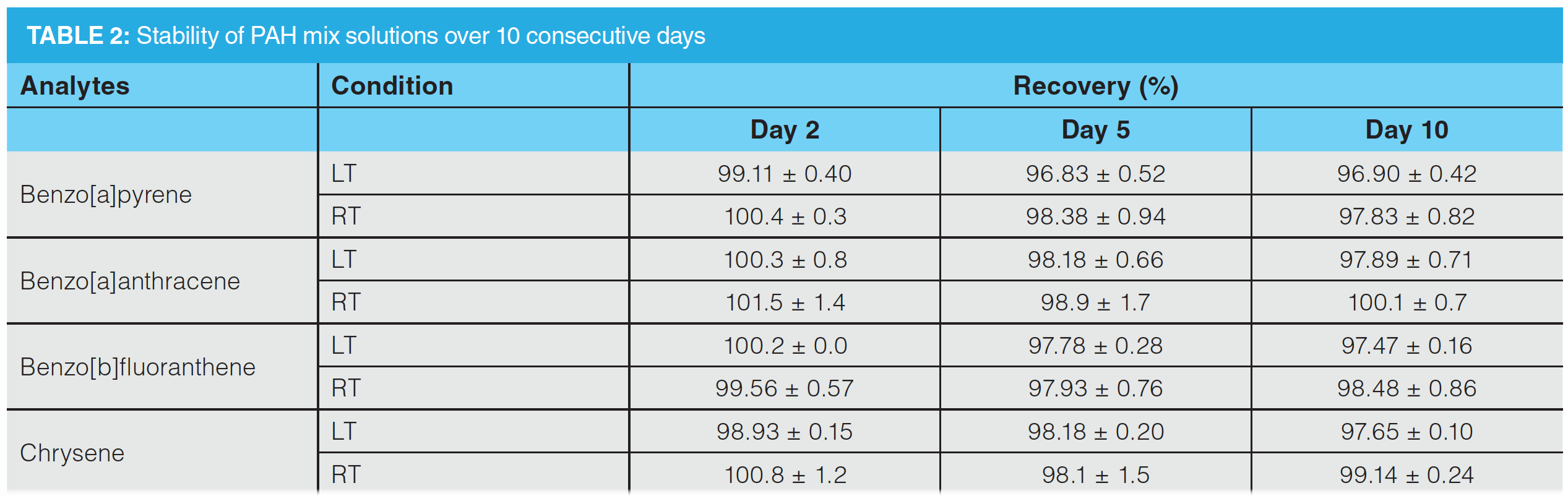
HPLC−FLD Method Validation: Important validation parameters were determined to highlight the applicability of the developed method. Individual results for all target analytes are summarized in Table 1. Analyte concentrations between 25% and 200% of target concentration showed excellent linearity with regression coefficients between 0.998 and 0.9996. The acquired values for LOD varied between 10.66 and 12.46 µg/L and LOQ values were obtained between 37.88 and 44.13 µg/L. In the overall method using SPE and a preconcentration factor of 10, these values correspond to a LOD of 1.66 and 1.25 µg/kg and a LOQ of 3.78 and 4.41 µg/kg plant material. Intra-day (n = 10) and inter-day (n = 30) repeatability demonstrated minimal differences, verified by RSD values below 1.04% for intra-day and 3.47% for inter-day repeatability. Stability of all proposed analytes in the standard mixture was recorded using HPLC–FLD. The mixture was measured over a time range of 10 days. Results showed that the concentration did not decrease below 96.90% ± 0.42%. Accuracy was determined at 50%, 100 %, and 150% of target concentration. Mean recoveries showed values ranging from 98.1% to 101.5%. RSDs ranged from 0.1% to 2.4%.


Stability Analyses: The analyses of the differently‑concentrated PAH standard mix solutions did not show a significant decrease of the analytes over the period of 10 days, neither at low temperature (LT) conditions nor at room temperature (RT) conditions. Representative for the stability of the PAH mix solutions, the observations on the stability of the 500 µg/L PAH mix at both LT and RT conditions are listed in Table 2.


Analyte Recovery Analysis: The use of filters did not show a significant difference in concentration of benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, or chrysene. But grinding of plant material, the evaporation for generating a dry extract, and the usage of different extraction vessel materials had a significant impact on analyte loss.
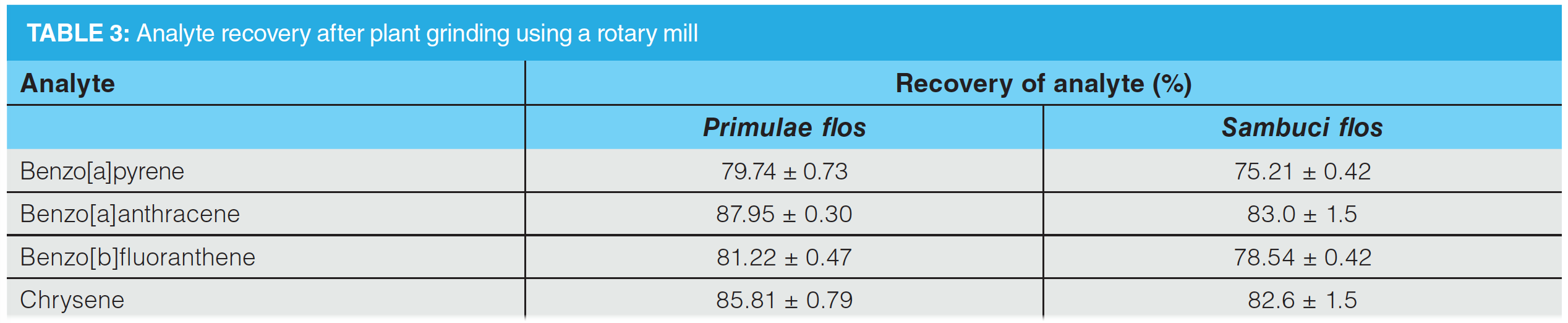
By using a rotary mill for plant grinding, the recovery of PAHs decreased from 88.0% to 75.2% (RSD < 1.6%). Cooling the samples using liquid nitrogen did not have a significant effect. Results for primulae flos and sambuci flos extracts are shown in Table 3.


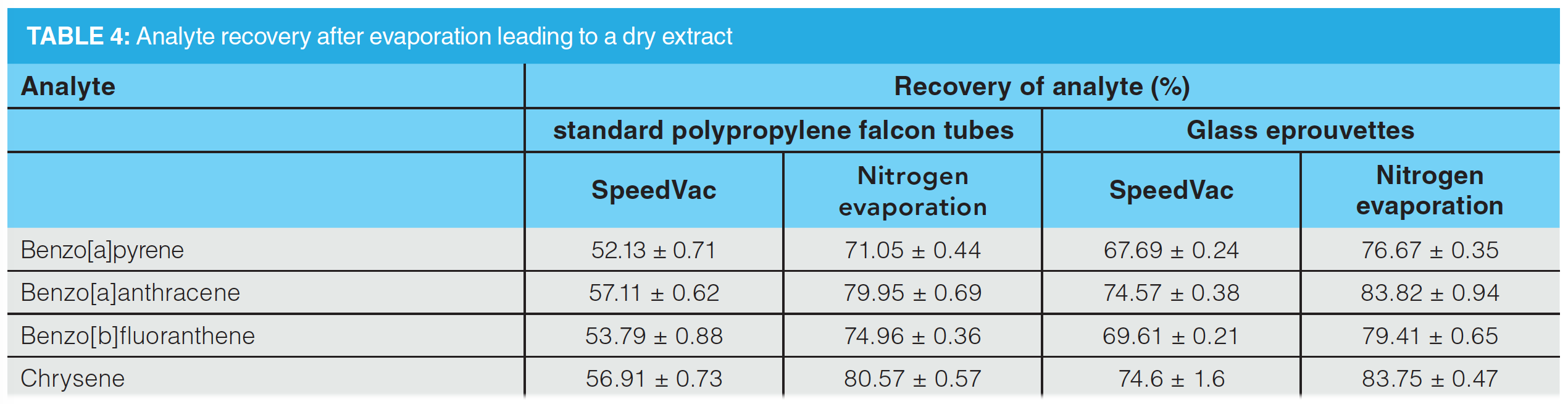
Evaporation using vacuum evaporation revealed recoveries between 74.6–52.1% (RSD < 1.7%), while evaporation using nitrogen evaporation showed significantly higher recovery between 83.8–71.1% (RSD < 1.0%). The usage of glass eprouvettes showed a significant improvement between 3–18%. Results are highlighted in Table 4.


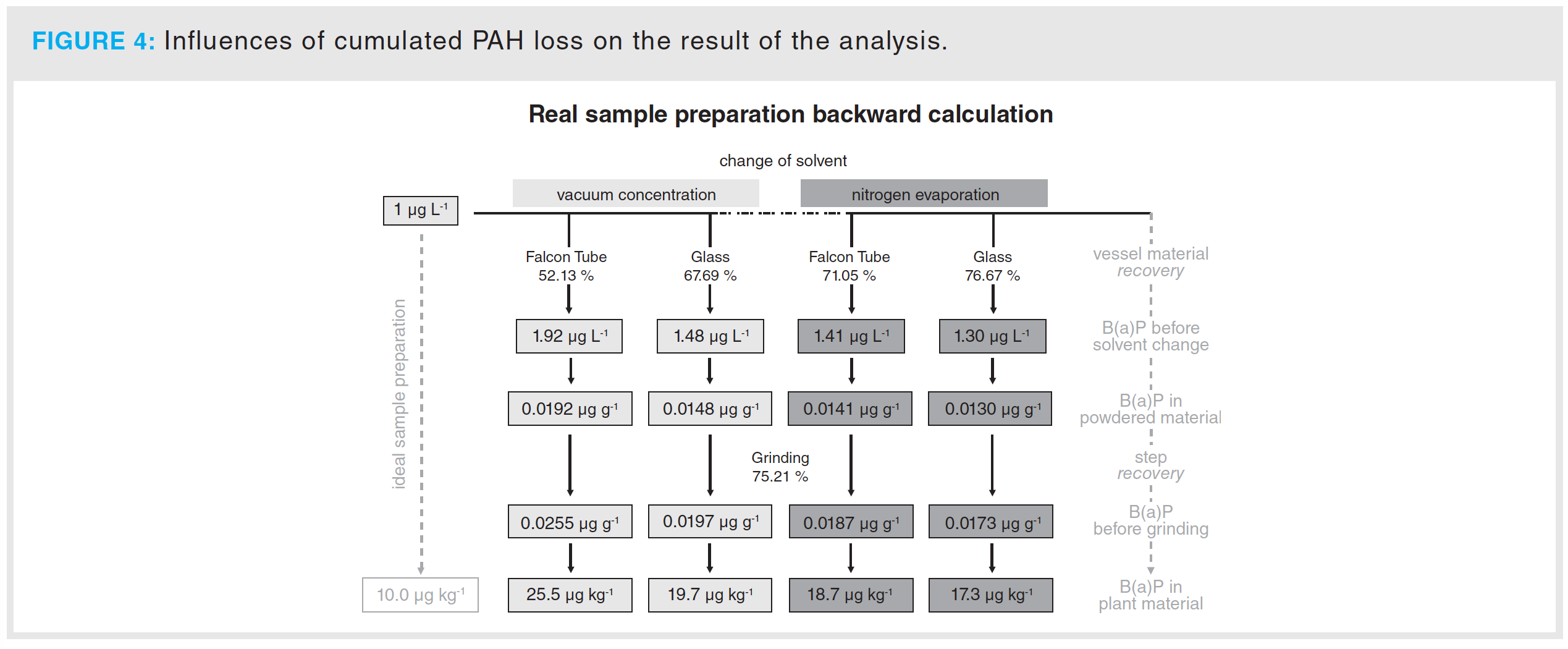
Calculation of the Deviation from the Theoretical True Value: Figure 4 shows a model calculation of the cumulated effect of the determined PAH losses during sample preparation. In the SPE procedure described by Stuppner et al. (1), a preconcentration factor of 10 was used. If contaminated plant material with a benzo[a]pyrene concentration of 1 µg/kg would be examined, 10 µg/L should be detected at the end of the analysis. In this model calculation, different evaporation techniques, including vacuum concentration and nitrogen evaporation, using various vial materials were used. As a result of the slight differences in the grinding step, the average recovery of the grinding was used. In the best case, when using a nitrogen evaporator with glass eprouvettes, a value of 17.3 µg/L was obtained, which results in a deviation of 73% from the theoretical true value. However, the worst case results in a deviation of 155% from the theoretical true value. Therefore, an analysis of PAH loss during extraction and pretreatment and the introduction of a correction factor is necessary.


Conclusion
In summary, the stability assessments of the standard solutions benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, and chrysene revealed no significant decrease or increase in the concentration over a period of ten consecutive days.
These findings are consistent with published literature. The main influence on PAH stability is photo degradation, and can be reduced by using amber glass vials (39). The use of PTFE and RC syringe filters did not show a significant influence either. Using a rotary mill for plant milling, the recovery rates of the PAHs were reduced to 88.0% and 75.2% (RSD < 1.6%). Cooling using liquid nitrogen did not improve the PAH loss either. Evaporation during solvent exchange showed the greatest influence, with polypropylene tubes and the vacuum evaporation method giving a recovery between 56.91–52.13% (RSD < 0.89%) for the investigated PAHs. These results do not match the existing literature. In literature a recovery of more than 90% was reported using 1-hexanol or 1-octanol as keeper substances. In addition, the samples were not evaporated to dryness to improve recovery, which is not practical for routine use (40). Using glass eprouvettes, the recovery values were improved from 67.7% to 74.6% (RSD < 1.6%). These results are consistent with the literature. Adsorption on polypropylene labware can also be overcome by liquid–liquid extraction (LLE) (40,41). Best results were obtained using a nitrogen evaporator with glass eprouvettes, with recovery rates varying between 76.67 and 83.82% (RSD < 0.95%). This is also in line with the existing literature. Rotary evaporation is described as a faster method with comparable recovery. However, nitrogen evaporation systems that can evaporate 72 samples in parallel offer benefits in terms of evaporation time and labour costs compared with rotary evaporation (40). The application of nitrogen vaporization has increased in recent publications and is therefore consistent with the results of this study (42). The model calculation (see “Calculation of the Deviation from the Theoretical True Value” section) also showed that nitrogen evaporation with glass vials produced the best results. However, an analysis of the PAH loss during extraction and pretreatment and the introduction of a correction factor are required.
The combination of these results with the findings published by Stuppner et al. (1) offers a valid method for PAH4 determination in plant matrix. In this study, a liquid handling robotic system was used in combination with an SPE-enrichment strategy using pre-loaded standards on poly(styrene-co-divinylbenzene) micro columns. For this purpose, the standards proposed by the EU PAH4 benzo[a]pyrene, benzo[a]anthracene, benzo[b]fluoranthene, and chrysene were used. The advantage is that the PAH standards can directly be pre-loaded by the manufacturer and the user does not have to prepare a standard solution or handle toxic standard substances. The PAHs are quantified using the standard addition method and 12 samples can be handled over a period of 17 min. Recoveries ranging from 90–103% (RSD < 8%) for PAH standards and 90–95% (RSD < 10%) for spiked plant extracts were achieved (1). The process is illustrated in Figure 5. The combination of the proposed method and the automatic enrichment strategy using pre-loaded SPE columns enables a quick and accurate method for determining PAH4 in herbal matrices such as primulae flos and sambuci flos.



Acknowledgements
The authors wish to highlight that Florian Meischl is recipient of a DOC Fellowship of the Austrian Academy of Sciences at the Institute of Analytical Chemistry and Radiochemistry, University of Innsbruck.
References
- S. Stuppner, S. Hussain, B. Märk, D. Gjerde, M. Rainer, T. Jakschitz, and G.K. Bonn, Anal. Methods 12(14), 1827–1833 (2020).
- R. Hänsel and O. Sticher, Eds., Pharmakognosie — Phytopharmazie (Springer-Lehrbuch, Berlin, 2010).
- A.N. Gennadiev and A.S. Tsibart, Euras. Soil Sci. 46(1), 28–36 (2013).
- B. Mohanty, P.R. Muduli, G. Cooper, S.K. Barik, D. Mahapatro, A.T. Behera, and A.K. Pattnaik, Bull. Env. Contam. & Toxicol. 99(1), 100–107 (2017).
- A. Paris, J. Ledauphin, P. Poinot, and J.-L. Gaillard, Env. Pollut. 234, 96–106 (2018).
- P. Benigni, R. Marin, K. Sandoval, P. Gardinali, and F. Fernandez‑Lima, J. Vis. Exp. 121, e55352 (2017). doi: 10.3791/55352.
- Kibblewhite, M., Env. Pollut. 242, 1331–1336 (2018).
- H.I. Abdel-Shafy and M.S.M. Mansour, Egyptian J. Petrol. 25(1), 107–123 (2016).
- Kommission, D.E., VERORDNUNG (EG) Nr. 1881/2006 Festsetzung der Höchstgehalte für bestimmte Kontaminanten in Lebensmitteln (2012).
- efsa, “Polycyclic Aromatic Hydrocarbons in Food – Scientific Opinion of the Panel on Contaminants in the Food Chain”, EFSA J., 724, 1–114 (2008). doi: 10.2903/j.efsa.2008.724.
- S. Danyi, F. Brose, C. Brasseur, Y.-J. Schneider, Y. Larondelle, L. Pussemier, J. Robbens, S. De Saeger, G. Maghuin-Rogister, and M.-L. Scippo, Anal. Chim. Acta. 633(2), 293–299 (2009).
- I. Windal, L. Boxus, and V. Hanot, J. Chrom. A. 1212(1–2), 16–22 (2008).
- Y. Shi, H. Wu, C. Wang, X. Guo, J. Du, and L. Du, Food Chem. 199, 75–80 (2016).
- T. Okuda, D. Naoi, M. Tenmoku, S. Tanaka, K. He, Y. Ma, F. Yang, Y. Lei, Y. Jia, and D. Zhang, Chemosphere 65(3), 427–435 (2006).
- D. Pan, J. Wang, C. Chen, C. Huang, Q. Cai, and S. Yao, Talanta 108, 117–122 (2013).
- Y. Wu, L. Xia, R. Chen, and B. Hu, Talanta 74(4), 470–477 (2008).
- M.R. Mannino and S. Orecchio, Atmos. Env. 42(8), 1801–1817 (2008).
- S. Orecchio, V.P. Ciotti, and L. Culotta, Food and Chem. Toxicol. 47(4), 819–826 (2009).
- B.S. Crimmins, and J.E. Baker, Atmos. Env. 40(35), 6764–6779 (2006).
- D. Asakawa, et al., Organohalogen Compd. 80, 45–48 (2008).
- T. Ghislain, P. Faure, and R. Michels, J. Am. Soc. Mass Spectrom. 23(3) 530–536 (2012).
- B. Jiang, Y. Liang, C. Xu, J Zhang, M. Hu, and Q. Shi, Env. Sci. Tech. 48(9), 4716–4723 (2014).
- H. Lim, T.M. Ahmed, C. Bergvall, and R. Westerholm, Anal. Bioanal. Chem. 409(24), 5619–5629 (2017).
- Gómez-Ruiz, J.Á. and T. Wenzl, Anal. Bioanal. Chem. 393(6), 1697–1707 (2009).
- P. Plaza-Bolaños, A.G. Frenich, and J.L.M. Vidal, J. Chrom. A. 1217(41), 6303–6326 (2010).
- M. Takino, S. Daishima, K. Yamaguchi, and T. Nakahara, J. Chrom. A. 928(1), 53–61 (2001).
- G.-W. Lien, C.-Y. Chen, and C.-F. Wu, Rapid Comms. Mass Spect. 21(22), 3694–3700 (2007).
- R. Luo and W. Schrader, Polycyclic Aromat. Compd. (2020). doi: 10.1080/10406638.2020.1748665.
- J. Hertzog, J., H. Naraoka, and P. Schmitt-Kopplin, Life (Basel) 9(2), 48 (2019).
- L. Hollosi and T. Wenzl, J. Chrom. A. 1218(1), 23–31 (2011).
- B. Pérez-Fernández, L. Viñas, and J. Bargiela, Env. Sci. Pollut. Res. Int. 25(16), 15862–15872 (2018).
- C.-D. Dong, C.-F. Chen, and C.-W. Chen, Int. J. Env. Res. Pub. Health 9(6), 2175–2188 (2012).
- E. Ballesteros, A. García Sánchez, and N. Ramos Martos, J. Chrom. A. 1111(1), 89–96 (2006).
- M. Suchanová, J. Hajšlová, M. Tomaniová, V. Kocourek, and L. Babička, J. Sci. Food and Agric. 88(8), 1307–1317 (2008).
- B. Janoszka, Food Chem. 126(3), 1344–1353 (2011).
- A. Farhadian, S. Jinap, F. Abas, and Z.I. Sakar, Food Control 21(5), 606–610 (2010).
- S.R. Gratz, L.A. Ciolino, A.S. Mohrhaus, B.M. Gamble, J.M. Gracie, D.S. Jackson, J.P. Roetting, H.A. McCauley, D.T. Heitkemper, F.L. Fricke, W.J. Krol, T.L Arsenault, J.C. White, M.M. Flottmeyer, and Y.S. Johnson, J. AOAC Int. 94(5), 1601–1616 (2011). doi: 10.5740/jaoacint.11-035.
- B. Veyrand, A. Brosseaud, L. Sarcher, V. Varlet, F. Monteau, P. Marchand, F. Andre, and B. Le Bizec, J. Chrom. A. 1149(2), 333–344 (2007).
- D. Dabrowska, A. Kot-Wasik, and J. Namiešnik, Polish J. Env. Studies 17(1), 17–24 (2008).
- C.C. Cheng, Polycyclic Arom.Comp. 23(3), 315–325 (2003).
- L. Wolska, M. Rawa-Adkonis, and J. Namieśnik, Anal. Bioanal. Chem. 382(6), 1389–1397 (2005).
- S.A.V. Tfouni, R.M. Reis, K. Kamikata, F.M.L. Gomes, M.A. Morgano, and R.P.Z. Furlani, Food Add. & Contam.: Part B 11(2), 146–152 (2018).
Stefan E. Stuppner received his M.Sc. in Biotechnology from MCI, Innsbruck, Austria, in 2017.He was awarded the title of engineer in 2015, by the Federal Ministry of Science, Research and Economy, Austria. He is currently doing his Ph.D. at the Institute of Analytical Chemistry and Radiochemistry at the University of Innsbruck, Austria. He specializes in new enrichment technologies, ambient mass spectrometry, and metabolomics.
Florian Meischl received his Ph.D. in Analytical Chemistry at the Institute of Analytical Chemistry and Radiochemistry, University of Innsbruck, Austria. His research focus is the development of new sorbents and methods for determination of pharmacological and toxicological relevant molecules. He received several fellowships and has published five articles as lead author.
Daniel Strolz received his B.Sc. in Chemistry at the Institute of Analytical Chemistry and Radiochemistry, from the University of Innsbruck, Austria, in 2019, and specializes in analytical research. He is currently doing his M.Sc. at the same university.
Sophia Mayr finished her studies in Chemistry in 2017. She is currently working towards her Ph.D. thesis at the Institute of Analytical Chemistry and Radiochemistry at the University of Innsbruck, Austria, with a focus on development of new methods for quality control and identification of pharmacological compounds in medicinal plants.
Shah Hussain is a scientist at ADSI—the Austrian Drug Screening Institute, GmbH, Innsbruck, Austria. He is a specialist in plant extractions, screening of active ingredients using state-of-the-art modern analytical instruments, solid-phase extraction and bio-analysis. He obtained his Ph.D. from the Institute of Analytical and Radiochemistry, University of Innsbruck, Austria in 2014.
Matthias Rainer is currently an associate professor at the Institute for Analytical Chemistry and Radiochemistry at the University of Innsbruck, Austria. The main focus of his research is the development of innovative analytical techniques using high-performance separation and enrichment methods in combination with mass spectrometry. Research fields comprise bioanalysis and phytoanalysis.
Christian Huck obtained his doctorate in chemistry in 1998 from the University in Innsbruck, Austria, where he continued to work as an assistant professor until the habilitation in 2006. In 2013, he received a call as a full professor to the University of Stuttgart, Germany, and in 2015, another call back to the University of Innsbruck, where he is currently vice-head of the Institute of Analytical Chemistry and Radiochemistry and head of the spectroscopy unit.
Thomas Jakschitz obtained his Ph.D. in analytical chemistry in 2010. He is currently employed as head of the analytical laboratories at the Austrian Drug Screening Institute GmbH. He is specialized in analysis of natural products, mainly in the topics phyto‑pharmacy, phyto-cosmetics and food supplements.
GŸnther K. Bonn studied chemistry at the University of Innsbruck and obtained his Ph.D. in 1979. In 1988 he spent time as a visiting professor at Yale University working with Prof. Csaba Horvath, one of the pioneers in modern HPLC. In 1996 he became Head of the Institute of Analytical Chemistry and Radiochemistry at the University of Innsbruck, Austria. He is currently CEO and Scientific Director of the Austrian Drug Screening Institute (ADS


![Stability and Recovery Influences of Benzo[a]pyrene, Benzo[a]anthracene, Benzo[b]fluoranthene, and Chrysene during Sample Preparation of Plant Matrices](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2Fb1144525ab2a1bd481ed407d90d9effacd96908d-1383x1041.png%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Stability and Recovery Influences of Benzo[a]pyrene, Benzo[a]anthracene, Benzo[b]fluoranthene, and Chrysene during Sample Preparation of Plant Matrices")


In the present study, a gradient reversed-phase high-performance liquid chromatography (RP-HPLC) method has been designed and validated to quantify ornidazole (OZ) in the marketed formulation (oral gel) with the application of QbD.


A column with chemically modified column hardware showed improvements in analytical performance for siRNA compared to a conventional stainless-steel column.




